Hemostasis
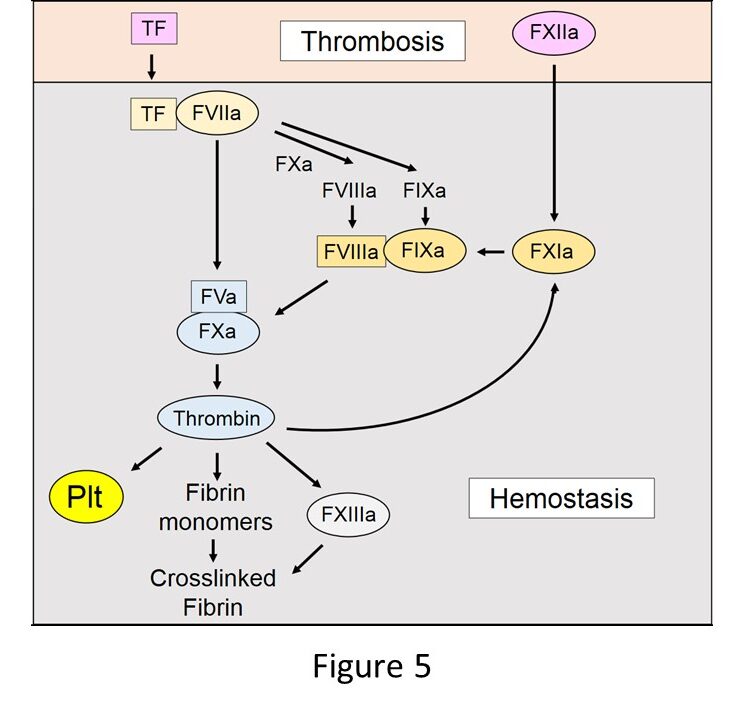
Components of the extrinsic pathway (TF, FVII, FV, FX, prothrombin) are essential for hemostasis (Figure 5). Gene knockout studies in mice show that an absence of any of these proteins led to universal lethality. In contrast, mice lacking FVIII or FIX can survive but exhibit increased bleeding after tail transection. These results indicate that FVIII and FIX are important for hemostasis but not essential. FVIII-/- and FIX-/- mice can be used as models of hemophilia A and B, respectively. The last piece of the hemostasis puzzle is FXI. Humans with a deficiency of FXI have a mild hemostasis defect that it often only reveal after a hemostatic challenge, such as tooth extraction. FXII does not contribute to hemostasis.
We generated mice that express low levels of TF (~1%) that have been useful in studying the role of TF in hemostasis. These mice exhibit a variety of spontaneous hemostatic defects in the uterus, placenta, lung, brain and testes. The hemostatic defects in low TF mice can be rescued by re-balancing the system by reducing the level of TFPI. Conversely, combining low levels of TF with a deficiency of PAR4 leads to an increase in fatal pulmonary hemorrhage.