
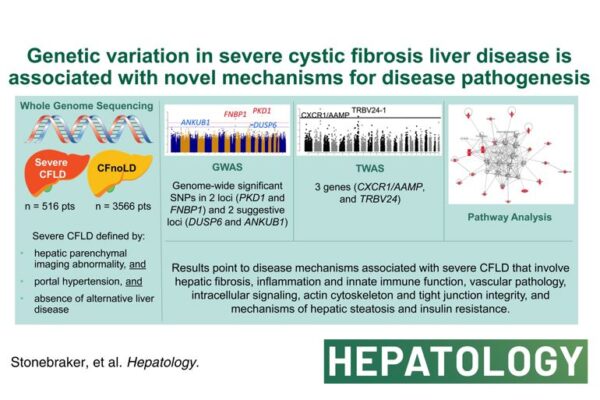
3 weeks ago


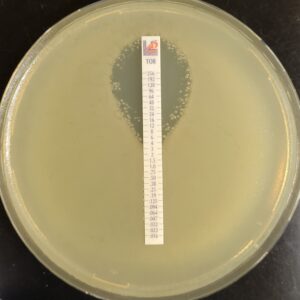
Antibiotic susceptibility testing for the antibiotic tobramycin (TOB) against Pseudomonas aeruginosa isolated from the sputum of a person with CF. The minimum inhibitory concentration of antibiotic is used both clinically and in research to understand how bacterial isolates will respond the antibiotic therapy. This image is from the Wolfgang Lab and was taken by Matthew Greenwald, a Graduate Student in the Department of Microbiology & Immunology.
3 weeks ago

4 weeks ago

1 month ago

2 months ago