Research
Some of our past work is summarized below:
- Regulation of DNA Polymerase kappa (Polk)
Polk is a DNA damage-tolerant and error-prone DNA polymerase that sustains DNA synthesis on templates containing damage (for example bulky DNA adducts from environmental carcinogens such as the combustion product Benzo[a]pyrene). Our work helped define the activation mechanism of Polk which is dependent upon mono-ubiquitination of the replication fork component PCNA (Bi et al., Mol Cell Biol 2004).
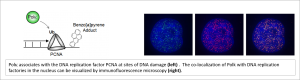
- Re-replication – a mechanism of genomic instability
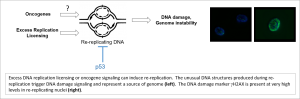
To maintain genome stability cells must replicate their genomes only once every cell cycle. We discovered that imbalanced expression of DNA
replication licensing factors such as CDT1 and CDC6 leads to re-replication – an unusual process whereby origins of DNA replication are activated multiple times each S-phase. Re-replicating cells accumulate excess DNA and acquire DNA damage. The human tumor suppressor protein p53 prevents re-replication (Vaziri et al., Mol Cell 2003). Interestingly, re-replication has also been proposed as a mechanism of oncogene-induced DNA damage.
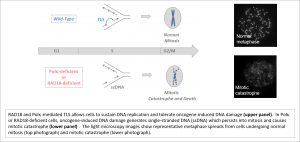
- DNA Polymerase kappa (Polk) allows tolerance of oncogene-induced DNA damage
Cancer cells experience high levels of DNA replication stress and DNA damage due to oncogene signaling. We found that Polk and its upstream activator RAD18 allow cells to tolerate oncogene-induced DNA damage (Yang et al., JCB 2017). Cells lacking Polk or RAD18 undergo fatal mitotic catastrophe in response to oncogene signaling. These results identify Polk and RAD18 as dependencies and molecular vulnerabilities of neoplastic cells.
- Cancer Testes Antigen (CTA)-mediated genome maintenance
Cancer Testes Antigens are germ cell-restricted proteins that are aberrantly expressed at high levels in many cancers. Increasingly it is suspected that CTAs confer tumorigenic phenotypes and contribute to carcinogenesis. Our proteomic screen for regulators of the TLS pathway identified Melanoma Antigen A4 (MAGE-A4, a CTA) as a novel RAD18-binding partner and activator (Gao et al., Nature Communications 2016). MAGE-A4-induced TLS provides a new mechanism for how neoplastic cells tolerate oncogenic and therapeutic DNA damage while simultaneously mutating their genomes via error-prone DNA replication.
