Research
Remarkable advances in supercomputing and artificial intelligence (AI) are transforming computational biology and chemistry in studies of molecules to cells. However, large gaps remain between the time scales of modern computer simulations (typically microseconds) and those of biological processes (milliseconds or longer). It has proven challenging to achieve sufficient sampling and compute thermodynamics and kinetics of biological systems, hindering effective drug design. Our research is focused on the development of novel theoretical and computational methods and AI techniques, which greatly enhance computer simulations and facilitate simulation analysis, and the application of these methods, making unprecedented contributions to biomolecular modeling and drug discovery. In collaboration with leading experimental groups, we combine complementary simulations and experiments to uncover functional mechanisms and design drugs of important biomolecules, including G-protein-coupled receptors (GPCRs), membrane-embedded proteases, RNA-binding proteins and RNA.
We are a highly interdisciplinary research lab. We work at the interface of computational biology, chemistry, molecular biophysics, bioinformatics and pharmacology. Our research aims to address three major topics as outlined below. But we always welcome innovative ideas and new collaborations, and as such our research program will keep evolving.
AI-driven drug discovery of G-protein-coupled receptors (GPCRs), membrane-embedded proteases, RNA-binding proteins, RNA and other important targets of shared interest with our experimental collaborators.
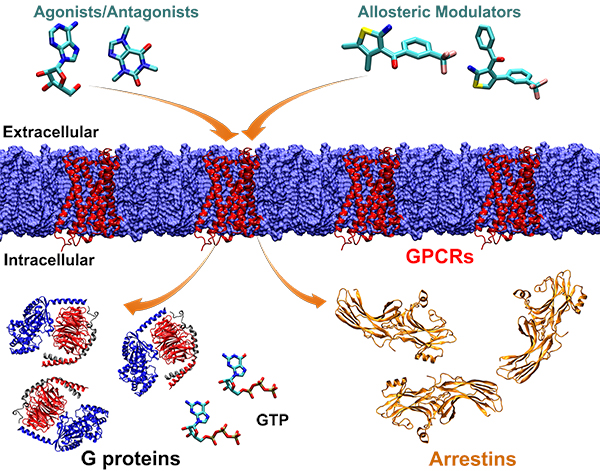
In particular, GPCRs are the largest family of human membrane proteins and represent primary targets of about one third of the currently marketed drugs. The traditional high-throughput screening of large compound libraries has become less attractive for drug discovery, due to increasingly high cost. GPCR drug discovery has suffered from major challenges, including the inherent flexibility of GPCRs and the toxicity of agonist and antagonist drugs that are designed to bind the conserved endogenous agonist-binding (“orthosteric”) site. It is appealing to develop selective therapeutics, such as “allosteric modulators” that modulate the function of target receptors by binding to diverse, topographically distant allosteric sites and “biased agonists” that activate specific downstream signaling pathways. Furthermore, it is extremely important to effectively account for GPCR flexibility in order to design potent drug molecules. To address these challenges, we will develop and apply state-of-the-art computational techniques (including all-atom accelerated molecular simulations, receptor ensemble docking, binding thermodynamics and kinetics calculations) to design selective drug leads of GPCRs and other important targets. Finally, we will collaborate with leading experimentalists to validate and optimize the lead compounds.
Development of biomolecular enhanced sampling and AI techniques.
Biomolecular recognition such as ligand binding to receptors, protein-peptide binding and protein-protein interactions (PPIs) plays key roles in cellular function. It is critical to accurately characterize biomolecular binding structures, thermodynamics and kinetics for effective therapeutic design. Building on a robust Gaussian accelerated molecular dynamics (GaMD) technique, the Miao Lab has developed innovative simulation methods, including “PeptiDock+GaMD” for improved modeling of protein-peptide binding structures19, Ligand GaMD (LiGaMD), Peptide GaMD (Pep-GaMD) and PPI-GaMD. LiGaMD, Pep-GaMD and PPI-GaMD, for the first time, enabled microsecond atomic simulations to capture repetitive dissociation and binding of small-molecule ligands, highly flexible peptides and proteins, respectively, thereby allowing for highly efficient and accurate calculations of ligand/peptide/protein binding free energies and kinetics.
In addition, we will integrate newly developed simulation algorithms with AI for further improved sampling and more efficient calculations biomolecular binding thermodynamics and kinetics, which are expected to facilitate computational drug discovery.
Multiscale modeling of cellular signaling pathways that are relevant to neurological disorders, heart failure and cancer.
It is of great biological and pharmaceutical interest to understand the signaling mechanisms of cells, especially those involving the GPCR drug targets. Specific questions include: What drives the binding of extracellular drugs/ligands (such as agonists, antagonists, and allosteric modulators) to GPCRs on the cell surface? How do activated GPCRs bind to specific intracellular proteins (e.g., G proteins and arrestins)? How do intracellular proteins recognize each other to activate complex signaling pathways? Answers to these questions will be valuable for developing effective treatments for human diseases that are caused by cellular signaling errors. Since the cellular signaling processes occur over a wide spectrum of atomic-to-subcellular scales, we will adopt a multiscale modeling strategy to investigate the pathways of cellular signaling.