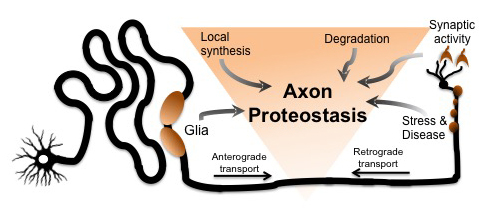
Dan Summers Ph.D. joined the department in 2005 performing his dissertation research in the lab of Doug Cyr Ph.D. He investigated protein quality pathways responsible for identifying misfolded or aggregated proteins, a process that is often debilitated in neurodegeneration. “My time in the Cyr lab was transformational… I developed the essential skills necessary to succeed as a scientist and the importance of digging deeper to solve fundamental questions of cell biology. I also developed a strong interest in protein homeostasis that still drives my research”. In 2011, Dan joined the labs of Jeff Milbrandt M.D. Ph.D and Aaron DiAntonio M.D. Ph.D. at Washington University in St. Louis. As a postdoctoral fellow, he characterized a new pathway responsible for the degeneration of injured axons in the nervous system. These studies revealed a new and exciting therapeutic target for many neurodegenerative disorders. In January 2019, Dan will begin a new position as Assistant Professor in the Department of Biology at the University of Iowa. “I wanted to combine my interest in protein homeostasis with the field of neuroscience in a unique way. If the axons in our body can grow over a meter in length, how do these subcellular compartments regulate protein quality control in complete isolation from the cell body? And how do these local proteostasis networks contribute to neurodegeneration?” Dan is grateful for his time at UNC, especially the stellar training environment in the Cyr laboratory and the entire department.
 You can learn more about Dan’s research interests here Link: biology.uiowa.edu/people/dan-summers
You can learn more about Dan’s research interests here Link: biology.uiowa.edu/people/dan-summers