

 |
**NEW**
High-Throughput Peptide Core and Arraying Facility
Brian D. Strahl, Ph.D. 3060 Genetic Medicine CB #7260 UNC School of Medicine Chapel Hill, NC 27599 Office: 919.843.3896 Lab: 919.843.3935 |
Background
Histone proteins play a central role in the packaging, organization and func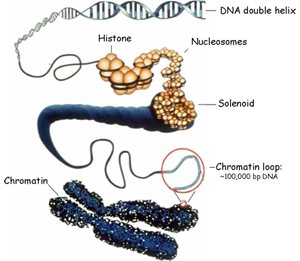 tion of our genome. These proteins, which include H3, H4, H2A and H2B, associate with DNA to form the fundamental unit of chromatin, the nucleosome. Along withthe actions of a linker histone called H1, strings of nucleosomes are further compacted into higher-order structures that are poorly defined but represent a barrier to genome access (Figure at Right). Thus, a significant question we and others are asking is how our genome is made accessible at the right place and time for all of the fundamental processes that need to occur in it such as gene expression, DNA repair and DNA replication.
tion of our genome. These proteins, which include H3, H4, H2A and H2B, associate with DNA to form the fundamental unit of chromatin, the nucleosome. Along withthe actions of a linker histone called H1, strings of nucleosomes are further compacted into higher-order structures that are poorly defined but represent a barrier to genome access (Figure at Right). Thus, a significant question we and others are asking is how our genome is made accessible at the right place and time for all of the fundamental processes that need to occur in it such as gene expression, DNA repair and DNA replication.
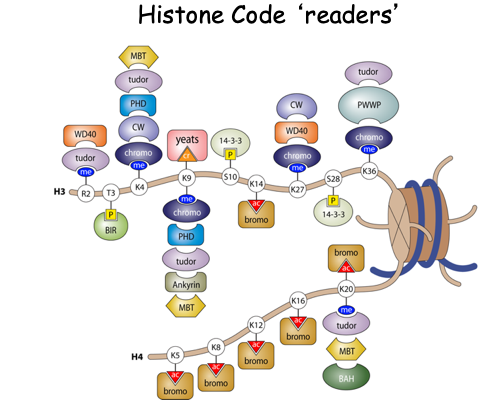
Histone post-translational modifications (PTMs) represent a major mechanism by which the structure and function of chromatin is regulated. Surprisingly, a vast number of covalent modifications, such as acetylation, methylation, ubiquitylation and phosphorylation exist on histones. Numerous studies indicate that these modifications work together in the form of a ‘histone code’ to regulate chromatin-based activities. This code functions, in part, through the direct recruitment of effector proteins utilizing specialized “reader” domains have been found to bind these histone PTMs (Figure below).
While much has been learned in the area of histone function, a great deal more work is needed to decipher how histone modifications impact chromatin structure and function. This area of work is extremely important, as numerous studies now show that a large number of proteins and enzymes that associate with chromatin (e.g., histone-modifiers, chromatin remodelers, and histone chaperones) are either recurrently mutated, deleted or overexpressed in many human diseases including cancer. In fact, even histone genes have been identified to be recurrently mutated in multiple cancers, leading to the realalization that there are "onco-histones".
In the Strahl lab, we are determining how the chromatin-modifying machinery “writes” and “reads” histone PTMs, and are defining how histone PTMs function in transc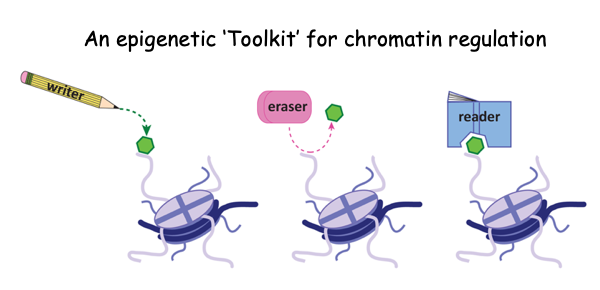 ription, DNA replication, and DNA repair. The collective functions of these writers, erasers and readers create an epigenetic "tool kit' that governs chromatin biology (Figure right). Our research can be devided more specifically into three, but overlapping, areas of investigation which include:
ription, DNA replication, and DNA repair. The collective functions of these writers, erasers and readers create an epigenetic "tool kit' that governs chromatin biology (Figure right). Our research can be devided more specifically into three, but overlapping, areas of investigation which include:
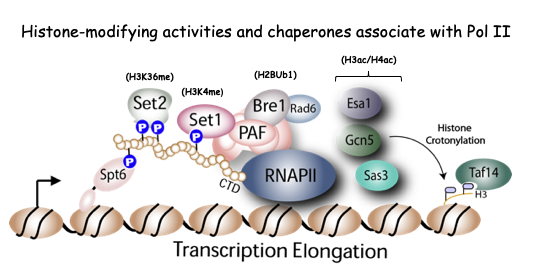
1) How do histone-modifying enzymes and chaperones contribute to transcription and maintenance of chromatin integrity? It is not precisely known how histone-modifying enzymes and histone chaperones promote proper gene transcription and direct processes such as DNA repair and the cell cycle. We have been addressing these challenges by focusing on Set2 (histone modifier) and Spt6 (histone chaperone) as models. Set2 suppresses inappropriate (“cryptic”) transcription; Spt6 mediates nucleosome reassembly during RNA Polymerase II (RNAPII) elongation. We recently discovered that Set2 methylation of histone H3 lysine36 (H3K36me) also regulates DNA repair and mRNA splicing. Similarly, we found an additional function of Spt6 by showing how it establishes RNAPII CTD phosphorylation during transcription elongation. Despite these advances, we have not fully determined the functions of Set2 and Spt6. Our lab is therefore examining two significant questions: 1) How does Set2-mediated H3K36 methylation contribute to transcription? And 2) How does Spt6 contribute to chromatin organization and genome integrity? (Figure below).
2) How do recently discovered histone PTMs contribute to gene transcription? Our lab played a key role in the discovery that YEATS domains selectively recognize histone crotonylation, a modification whose function is largely unknown. Significantly, lysine crotonylation represents a series of possible “acylations” (e.g., acetylation, propionylation and butyrylation) derived from different acyl-CoA forms generated by metabolism during nutrient flux However, we know little mechanistically about how these acylations control transcription and how their dysregulation contributes to cancer progression.
We are addressing this knowledge gap by examining the occurrence and fate of histone acetylation across the yeast metabolic cycle (YMC) – a well-established cycle in which, under fixed and low-glucose conditions, cells oscillate through states of high oxygen consumption (HOC; i.e., oxidative metabolism) and low oxygen consumption (LOC; i.e., reductive metabolism). The metabolic pathways activated or repressed in each YMC phase are governed by discrete, well documented transcription programs, and, further, histone acetylation levels oscillate across the YMC (with the highest levels occurring in the HOC phase in which the TCA cycle generates abundant acetyl-CoA). With this system, we are determining how metabolism contributes to the establishment of these newly discovered chromatin marks and how YEATS domain-containing proteins read these acylations on histones to regulate transcription.
3) To what extent do histone PTMs work together to regulate downstream events in chromatin? In 2000, Dr. C. David Allis and I put forward the a ‘histone code’ hypothesis to stimulate new thinking about the functions of histone PTMs. We envisioned that, besides histone PTMs directly affecting chromatin structure, these PTMs also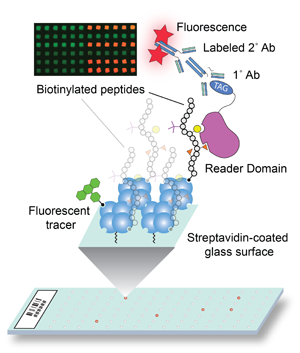 selectively recruit, either singly or in combination, effector proteins (readers) that direct specific downstream events in chromatin. Since this original postulate, numerous domains have been identified that interact with histones and/or their PTMs. Still, we do not know how these domains are influenced by neighboring PTMs (cross-talk), and, further, we do not know how paired domains and domains that occur in multiple proteins within a macromolecular complex (e.g., SWI/SNF) contribute to combinatorial histone PTM readout. Thus, we are examining the molecular basis of chromatin engagement by effector proteins and how multivalent interactions regulate genome access and function. Towards this goal, we developed a next generation, high-density histone peptide microarray platform to measure the histone interactions of known and putative histone readers, paired reader domains, and the substrate specificities of histone-modifying enzymes. This array platform is enabling us to characterize countless reader domains and define the molecular underpinnings of histone PTM interaction (Figure above).
selectively recruit, either singly or in combination, effector proteins (readers) that direct specific downstream events in chromatin. Since this original postulate, numerous domains have been identified that interact with histones and/or their PTMs. Still, we do not know how these domains are influenced by neighboring PTMs (cross-talk), and, further, we do not know how paired domains and domains that occur in multiple proteins within a macromolecular complex (e.g., SWI/SNF) contribute to combinatorial histone PTM readout. Thus, we are examining the molecular basis of chromatin engagement by effector proteins and how multivalent interactions regulate genome access and function. Towards this goal, we developed a next generation, high-density histone peptide microarray platform to measure the histone interactions of known and putative histone readers, paired reader domains, and the substrate specificities of histone-modifying enzymes. This array platform is enabling us to characterize countless reader domains and define the molecular underpinnings of histone PTM interaction (Figure above).
Additional collaborative projects
The Strahl lab is also collaborating with a variety of labs at UNC and beyond to understand chromatin regulation and epigenetics.
One area of focus is on the control of epigenetics using optognetic approaches. In collaboration with Dr. Brian Kuhlman's lab at UNC, we have been developing new tools to target, on demand, epigenetic regulators from the nucleus to the cytoplasm (and vise versa). This system, which involves a light-inducible control switch fused to chromatin regulators, will allow us to ask new questions about the kinetics of histone methylation distribution and removal in relation to transcription. We are creating light-inducible control switches for a number of chromatin regulators.
Another collaborative example is with the development of a new platform to study histone modifications in Drosophila. In collaboration with Greg Matera, Bob Duronio and Dan McKay, a histone replacement system has been established where we can introduce histone point mutations to study key biological processes such as heterochromatic gene silencing. A paper describing this platform is now accepted in Developmental Cell (McKay et al. 2015).
