Long Read Technologies
Oxford Nanopore
 At the High Throughput Sequencing Center, our goal is to provide the research community at UNC with access to cutting edge sequencing platforms. To that end, we are offering Oxford Nanopore Sequencing on the GridION platform.
At the High Throughput Sequencing Center, our goal is to provide the research community at UNC with access to cutting edge sequencing platforms. To that end, we are offering Oxford Nanopore Sequencing on the GridION platform.
Oxford Nanopore Technologies offers direct sequencing of native DNA or RNA, or samples that have been amplified with PCR. Nanopore sequencing can read any length of DNA/RNA, from short to ultra-long. It is useful for applications such as De novo assembly, scaffolding and finishing, bridging repetitive regions, structural variation, SNVs and phasing, targeted sequencing, RNA analysis, metagenomics, and epigenetics.
- A single MinION flow cell can produce reads up to 2 Mb long, and provide up to 10 Gb of sequencing data
- The GridION can run up to 5 MinION flow cells at one time
- There is no fixed run time; a user can run any of the systems for a short or long period of time, as data is streamed in real time
The Oxford Nanopore’s unique direct molecule sequencing platform is based on protein nanopores set in a polymer membrane. Current is passed through the nanopore, and as DNA/RNA is passed through the pore, a disruption in current is detected.
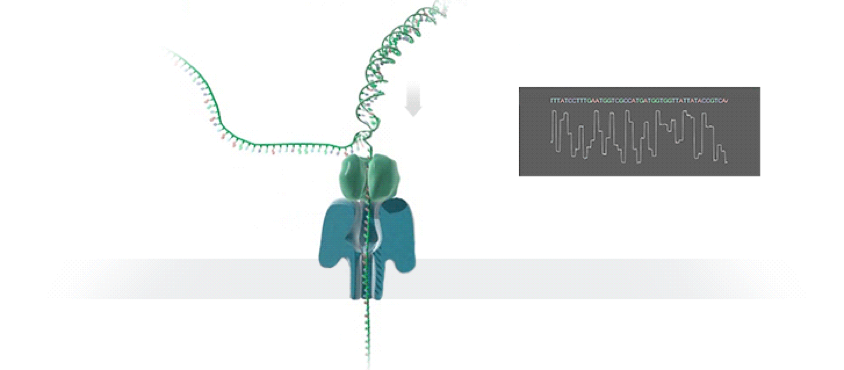
DNA Sequencing (from nanoporetech.com): A strand of DNA is passed through a nanopore. The current is changed as the bases G, A, T and C pass through the pore in different combinations.
Oxford Nanopore can produce ultra-long read sequencing reads, limited by DNA length/quality. In our hands we regularly generate reads >100kb. There are a host of applications, including:
- Rapid sequence identification
- Utilizing long reads for improved genome assembly
- Analysis of full length RNA transcripts from cDNA (PCR and PCR-free)
- Direct sequencing of RNA molecules
- Metagenomic analysis
- Structural variant detection
- Copy Number detection in complex regions
To learn more about Oxford Nanopore sequencing at the HTSF, read our white paper or contact Piotr Mieczkowski at Piotr_Mieczkowski@med.unc.edu or Tara Skelly at tskelly@email.unc.edu.
PacBio Sequel
For modest projects, the HTSF has a collaboration with UNC’s Vironomics Core to use their PacBio Sequel IIe long-read sequencer. (Larger project can be supported by our partner institutions). The PacBio Sequel IIe instrument has the ability to generate reads between 6,000 and 20,000 base pairs, enabling the generation of complete genomes rather than limited exome sequences.For more information, see the Vironomics Core website
The PacBio system utilizes Single Molecule, Real-Time (SMRT) technology, eliminating the need for amplification. The DNA submitted serves as a direct template for the sequencing reaction via the ligation of hairpin adapters that create a circular template. Real-time sequencing occurs continuously across this circular template, producing extra-long read lengths and high consensus accuracy. These advantages make the PacBio Sequel a powerful tool in accurately calling SNPs and sequencing through CG-rich areas.
The PacBio Sequel produces up to 30 kb sequence reads with up to 7X more data output than the RS II. The Sequel System is ideal for rapidly and cost-effectively generating high-quality whole genome de novo assemblies, and facilitating full-length transcriptome sequencing (IsoSeq) efforts. Typical data output per SMRT Cell is 5-8 Gb.
For more information, see the PacBio website
For small projects, please contact Dr. Dittmer for project questions. For larger projects, please contact HTSF about high volume options at NCSU and Duke.
Contact Vironomics Core
https://www.med.unc.edu/vironomics/
vironomics@med.unc.edu OR
dirk_dittmer@med.unc.edu
(919)843-5292
Contact HTSF
htsf@med.unc.edu
(919)966-3763