Specialty Areas:
Chronology:
- 1997
- Ph.D. (summa cum laude) Cell Biology: Protein Processing, University of Regensburg, Germany
- 1997-1998
- Postdoctoral Fellow, Laboratory of Dr. W. Tanner, Institute of Cell Biology, University of Regensburg, Germany
- 1998-2001
- Postdoctoral Research Fellow, Laboratory of Dr. J.R. Riordan, Mayo Clinic, Scottsdale, AZ
- 2001-2005
- Research Associate, Mayo Clinic College of Medicine, Scottsdale, AZ
- 2005-2012
- Assistant Professor of Cell and Developmental Biology, UNC, Chapel Hill, NC
- 2012-2015
- Director, Correction Core (NIH MTCC), Cystic Fibrosis/Pulmonary Research & Treatment Center, UNC
- 2012-2016
- Assistant Professor of Cell Biology and Physiology, UNC
- 2015-Present
- Director, CFTR Functional Analysis Core (CFF RDP), Marsico Lung Institute (MLI), UNC
- 2015-Present
- Director, Cystic Fibrosis Molecular/Functional Measurement Pre-Clinical Core (NIH CFRTCC) MLI, UNC
- 2016-2020
- Associate Professor of Cell Biology and Physiology, UNC
- 2020-2026
- Associate Professor of Pediatrics, Pediatric Pulmonology, UNC
- 2020-Present
- Adjunct Associate Professor of Cell Biology and Physiology, UNC
- 2026-Present
- Professor of Pediatrics, Pediatric Pulmonology, UNC
Research Focus:
A major goal of my studies is to uncover potential targets for therapy of chronic lung diseases, including cystic fibrosis (CF).
Specifically, I work on pharmacological modulation of airway epithelial ion channels, such as the CF transmembrane conductance regulator (CFTR) that is mutated in CF, the epithelial sodium channel (ENaC), and calcium-activated chloride channels (CaCCs) in physiologically relevant model systems. In particular, my research focuses on CFTR processing, intracellular trafficking, and rescue. My laboratory is interested in improving pre-clinical models for CFTR research and drug discovery in CF, and we have developed reliable methodology to analyze ion channel physiology in relevant primary human bronchial epithelial (HBE), human nasal epithelial (HNE) and GI cultures. We are applying these techniques and electrophysiological assays to evaluate CFTR rescue by small-molecule compounds in primary and conditionally reprogrammed epithelial nasal, bronchial, and intestinal cultures and tissues. In addition, we have developed and are utilizing state-of-the-art pre-clinical assays to investigate the efficacy of novel therapeutic approaches to CF in relevant in vitro, ex vivo, and in vivo models of CF including organoids from different tissues.
More recently, my research has been investigating how bacterial and viral infection and inflammation affect ion channel function and protein interactions in airway epithelia. Furthermore, we are developing novel precision medicine models for lung diseases beyond CF. Recently, we have utilized our ion channel expertise to investigate properties of the SARS-CoV-2 envelope viroporin. This viral ion channel plays a crucial role in SARS-CoV-2 pathogenesis and is being assessed as a target for development of COVID-19 therapeutics.
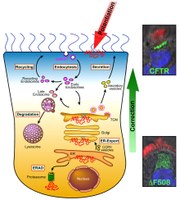
Fig. 1. Rescue of CFTR. The mutant CFTR protein, F508del, is retained at the ER in differentiated primary human bronchial epithelial (HBE) cells. Corrector compounds promote transfer of mutant CFTR from the ER to the apical membrane, whereas potentiator compounds increase activity of CFTR at the apical surface. The insets show confocal immunofluorescence of CFTR (green) expressed in primary HBE cells. F508del CFTR is retained within the cell (bottom panel) while wild-type CFTR is at the cell surface (top panel). Cilia are stained in red and nuclei in blue.

Fig. 2. Correction of mutant CFTR in CF HBE cells (P67L/∆F508).Short-circuit currents were measured in Ussing chambers. Amiloride was added to inhibit ENaC, forskolin to stimulate CFTR, VX-770 to potentiate CFTR, and CFTR inhibitor-172 to inhibit CFTR. The blue stripe, treatment with VX-809 (5 µM) + VX-770 (1 µM), resulted in the highest level of CFTR function for this particular mutant.
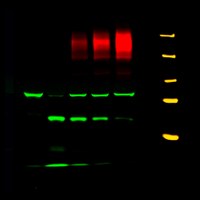
Fig. 3. CFTR impedes proteolysis of ENaC. Expression of increasing amounts of CFTR (shown in red) diminishes appearance of matriptase-induced proteolytic fragments of α-ENaC (shown in green). CFTR, ENaC, and matriptase were co-expressed in Xenopus oocytes. Molecular weight markers (250, 150, 100, 75 and 50 kDa) are shown in yellow.
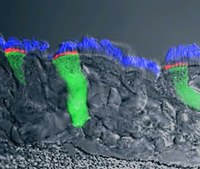
Fig. 4. Virally delivered expression of CFTR in epithelial cells. Wild-type CFTR resides at the apical membrane in primary HBE cells. Cilia are stained in blue, virally delivered CFTR-expressing cells in green, and CFTR is shown in red.
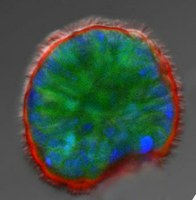
Fig. 5. Human primary nasal cells grown as 3D sphere cultures. Nasal epithelial cells obtained from individuals can be cultured into 3D spheres and used for studying ion channel function. Apical membrane and cilia are stained in red, cells in green, and nuclei in blue.
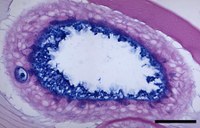
Fig. 6. Human primary bronchial sphere grown in matrigel. Bronchial cells obtained from individuals can be cultured into 3D spheres for studying ion channel function. When grown in matrigel, the apical surface faces the inside of the sphere. This section is stained with AB PAS. Bar=50µm.
Selected Bibliography:
- Yeshwante S, Mahadevan R, Sule O, Tiley JB, Luterbach CL, McCann S, Fallon JK, Smith PC, Tan ML, Walenga R, Newman B, Zhao L, Gentzsch M, Esther CR Jr, Rao GG. Modeling lung transporters and their influence on inhaled drug disposition. Pharmacol Rev. 2026 Jan;78(1):100100. doi: 10.1016/j.pharmr.2025.100100. PMID: 41297206.
- Cholon DM, Aleksandrov LA, Quinney NL, Boyles SE, Jensen TJ, Aleksandrov AA, Gentzsch M. Implications for cystic fibrosis therapy: Potentiator icenticaftor is superior to ivacaftor in improving function and maintaining stability of F508del CFTR. Sci Prog. 2025 Oct-Dec;108(4):368504251384892. doi: 10.1177/00368504251384892. Epub 2025 Oct 7. PMID: 41055920; PMCID: PMC12508541.
- Gentzsch M. Pulmonary Ionocytes at the Crossroads of Absorption and Secretion: Clues from the Ferret. Am J Respir Crit Care Med. 2025 Oct;211(10):1898-1900. doi: 10.1164/rccm.202508-2001ED. PMID: 40920899. PMCID: PMC12555049.
- Sun L, Walls SA, Dang H, Quinney NL, Sears PR, Sadritabrizi T, Hasegawa K, Okuda K, Asakura T, Chang X, Zheng M, Mikami Y, Dizmond FU, Danilova D, Zhou L, Deshmukh A, Cholon DM, Radicioni G, Rogers TD, Kissner WJ, Markovetz MR, Guhr Lee TN, Gutay MI, Esther CR Jr, Chua M, Grubb BR, Ehre C, Kesimer M, Hill DB, Ostrowski LE, Button B, Gentzsch M, Robinson C, Olivier KN, Freeman AF, Randell SH, Vladar E, O’Neal WK, Boucher RC Jr, Chen G. STAT3-dependent Regulation of CFTR and Ciliogenesis Is Essential for Mucociliary Clearance and Innate Airway Defense in Hyper-IgE Syndrome. Am J Respir Crit Care Med. 2025 Oct;211(10):1951-1969. doi: 10.1164/rccm.202407-1415OC. PMID: 40315437. PMCID: PMC12555045.
- Bourassa MH, Sillon G, Ding S, Chioccioli M, Lek M, Ma K, Mejia-Garcia A, Gravel S, Vinh DC, Knowles MR, Leigh MW, Davis SD, Ferkol T, Olivier KN, Schecterman EN, Yin W, Sears PR, Gentzsch M, Boyles SE, Bennett WD, Zeman KL, Ostrowski LE, Zariwala MA, Shapiro AJ. ODAD4-Related Primary Ciliary Dyskinesia: Report of Five Cases and a Founder Variant in Quebec. Cells. 2025 Sep 18;14(18):1460. doi: 10.3390/cells14181460. PMID: 41002425; PMCID: PMC12468610.
- Gentzsch M, Esther CR Jr, Ribeiro CMP. Airway inflammation enhances the effectiveness of elexacaftor-tezacaftor-ivacaftor therapy for cystic fibrosis and CFTRN1303K mutation. Lancet Respir Med. 2024 Nov;12(11):e67. doi: 10.1016/S2213-2600(24)00305-9. PMID: 39395437.
- Okuda K, Gentzsch M. Pulmonary Ionocytes: What Are They Transporting and Which Way? Am J Respir Crit Care Med.2024 Sep 15;210(6):705-707. doi: 10.1164/rccm.202404-0727ED. PMID: 38701428. PMCID: PMC11418888.
- Gentzsch M, Baker B, Cholon DM, Kam CW, McKinzie CJ, Despotes KA, Boyles SE, Quinney NL, Esther CR Jr, Ribeiro CMP. Cystic fibrosis airway inflammation enables elexacaftor/tezacaftor/ivacaftor-mediated rescue of N1303K CFTR mutation. ERJ Open Res. 2024 Jan 15;10(1):00746-2023. doi: 10.1183/23120541.00746-2023. PMID: 38226069; PMCID: PMC10789252.
- Cholon DM, Greenwald MA, Higgs MG, Quinney NL, Boyles SE, Meinig SL, Minges JT, Chaubal A, Tarran R, Ribeiro CMP, Wolfgang MC, Gentzsch M. A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection. Cells. 2023 Nov 13;12(22):2618. doi: 10.3390/cells12222618. PMID: 37998353; PMCID: PMC10670530.
- Mikami Y, Grubb BR, Rogers TD, Dang H, Asakura T, Kota P, Gilmore RC, Okuda K, Morton LC, Sun L, Chen G, Wykoff JA, Ehre C, Vilar J, van Heusden C, Livraghi-Butrico A, Gentzsch M, Button B, Stutts MJ, Randell SH, O’Neal WK, Boucher RC. Chronic airway epithelial hypoxia exacerbates injury in muco-obstructive lung disease through mucus hyperconcentration. Sci Transl Med. 2023 Jun 7;15(699):eabo7728. doi: 10.1126/scitranslmed.abo7728. PMID: 37285404. PMCID: PMC10664029.
- Ramalho AS, Amato F, Gentzsch M. Patient-derived cell models for personalized medicine approaches in cystic fibrosis. J Cyst Fibros. 2023 Mar;22 Suppl 1(Suppl 1):S32-S38. doi: 10.1016/j.jcf.2022.11.007. PMID: 36529661. PMCID: PMC9992303.
- Ribeiro CMP, Gentzsch M. Editorial overview – 2022 respiratory issue: Cystic fibrosis pathophysiology, models, and novel therapies. Curr Opin Pharmacol. 2022 Dec;67:102289. doi: 10.1016/j.coph.2022.102289. PMID: 36152600.
- Ruan J, Liang D, Yan W, Zhong Y, Talley DC, Rai G, Tao D, LeClair CA, Simeonov A, Zhang Y, Chen F, Quinney NL, Boyles SE, Cholon DM, Gentzsch M, Henderson MJ, Xue F, Fang S. A small-molecule inhibitor and degrader of the RNF5 ubiquitin ligase. Mol Biol Cell. 2022 Nov 1;33(13):ar120. doi: 10.1091/mbc.E22-06-0233. PMID: 36074076. PMCID: PMC9634977.
- Lee RE, Lewis CA, He L, Bulik-Sullivan EC, Gallant SC, Mascenik TM, Dang H, Cholon DM, Gentzsch M, Morton LC, Minges JT, Theile JW, Castle NA, Knowles MR, Kimple AJ, Randell SH. Small-molecule eRF3a degraders rescue CFTR nonsense mutations by promoting premature termination codon readthrough. J Clin Invest. 2022 Sep 15;132(18):e154571. doi: 10.1172/JCI154571. PMID: 35900863; PMCID: PMC9479597.
- Harrison NL, Abbott GW, Gentzsch M, Aleksandrov A, Moroni A, Thiel G, Grant S, Nichols CG, Lester HA, Hartel A, Shepard K, Garcia DC, Yazawa M. How many SARS-CoV-2 “viroporins” are really ion channels? Commun Biol. 2022 Aug 25;5(1):859. doi: 10.1038/s42003-022-03669-2. PMID: 36008538; PMCID: PMC9411608.
- Cholon DM, Gentzsch M. Established and novel human translational models to advance cystic fibrosis research, drug discovery, and optimize CFTR-targeting therapeutics. Curr Opin Pharmacol. 2022 Jun;64:102210. doi: 10.1016/j.coph.2022.102210. PMID: 35462105. PMCID: PMC9167699.
- Ghigo A, Murabito A, Sala V, Pisano AR, Bertolini S, Gianotti A, Caci E, Montresor A, Premchandar A, Pirozzi F, Ren K, Della Sala A, Mergiotti M, Richter W, de Poel E, Matthey M, Caldrer S, Cardone RA, Civiletti F, Costamagna A, Quinney NL, Butnarasu C, Visentin S, Ruggiero MR, Baroni S, Crich SG, Ramel D, Laffargue M, Tocchetti CG, Levi R, Conti M, Lu XY, Melotti P, Sorio C, De Rose V, Facchinetti F, Fanelli V, Wenzel D, Fleischmann BK, Mall MA, Beekman J, Laudanna C, Gentzsch M, Lukacs GL, Pedemonte N, Hirsch E. A PI3Kγ mimetic peptide triggers CFTR gating, bronchodilation, and reduced inflammation in obstructive airway diseases. Sci Transl Med. 2022 Mar 30;14(638):eabl6328. doi: 10.1126/scitranslmed.abl6328. PMID: 35353541. PMCID: PMC9869178.
- Morrison CB, Shaffer KM, Araba KC, Markovetz MR, Wykoff JA, Quinney NL, Hao S, Delion MF, Flen AL, Morton LC, Liao J, Hill DB, Drumm ML, O’Neal WK, Kesimer M, Gentzsch M, Ehre C. Treatment of cystic fibrosis airway cells with CFTR modulators reverses aberrant mucus properties via hydration. Eur Respir J. 2022 Feb 3;59(2):2100185. doi: 10.1183/13993003.00185-2021. PMID: 34172469. PMCID: PMC8859811.
- Farinha CM, Gentzsch M. Revisiting CFTR Interactions: Old Partners and New Players. Int J Mol Sci. 2021 Dec 7;22(24):13196. doi: 10.3390/ijms222413196. PMID: 34947992; PMCID: PMC8703571.
- Ribeiro CMP, Gentzsch M. Impact of Airway Inflammation on the Efficacy of CFTR Modulators. Cells. 2021 Nov 22;10(11):3260. doi: 10.3390/cells10113260. PMID: 34831482; PMCID: PMC8619863.
- Dang Y, van Heusden C, Nickerson V, Chung F, Wang Y, Quinney NL, Gentzsch M, Randell SH, Moulton HM, Kole R, Ni A, Juliano RL, Kreda SM. Enhanced delivery of peptide-morpholino oligonucleotides with a small molecule to correct splicing defects in the lung. Nucleic Acids Res. 2021 Jun 21;49(11):6100-6113. doi: 10.1093/nar/gkab488. PMID: 34107015; PMCID: PMC8216463.
- Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, Chen G, Kato T, Asakura T, Gilmore RC, Morton LC, Lee RE, Mascenik T, Yin WN, Barbosa Cardenas SM, O’Neal YK, Minnick CE, Chua M, Quinney NL, Gentzsch M, Anderson CW, Ghio A, Matsui H, Nagase T, Ostrowski LE, Grubb BR, Olsen JC, Randell SH, Stripp BR, Tata PR, O’Neal WK, Boucher RC. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am J Respir Crit Care Med. 2021 May 15;203(10):1275-1289. PMID: 33321047. PMCID: PMC8456462.
- He L, Kennedy AS, Houck S, Aleksandrov A, Quinney NL, Cyr-Scully A, Cholon DM, Gentzsch M, Randell SH, Ren HY, Cyr DM. DNAJB12 and Hsp70 triage arrested intermediates of N1303K-CFTR for endoplasmic reticulum-associated autophagy. Mol Biol Cell. 2021 Apr 1;32(7):538-553. PMID: 33534640; PMCID: PMC8101465.
- Gentzsch M, Cholon DM, Quinney NL, Martino MEB, Minges JT, Boyles SE, Guhr Lee TN, Esther CR Jr, Ribeiro CMP. Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies. Front Pharmacol. 2021 Mar 30;12:628722. PMID: 33859562; PMCID: PMC8042279.
- Xu J, Livraghi-Butrico A, Hou X, Rajagopalan C, Zhang J, Song J, Jiang H, Wei HG, Wang H, Bouhamdan M, Ruan J, Yang D, Qiu Y, Youming X, Barrett RP, McClellan SA, Mou H, Wu Q, Chen X, Rogers TD, Wilkinson KJ, Gilmore RC, Esther CR Jr, Zaman K, Liang X, Sobolic M, Hazlett L, Zhang K, Frizzell RA, Gentzsch M, O’Neal WK, Grubb BR, Chen YE, Boucher RC, Sun F. Phenotypes of CF rabbits generated by CRISPR/Cas9-mediated disruption of the CFTR gene. JCI Insight. 2021 Jan 11;6(1):e139813. PMID: 33232302; PMCID: PMC7821608.
- Gentzsch M, Rossier BC. A Pathophysiological Model for COVID-19: Critical Importance of Transepithelial Sodium Transport upon Airway Infection. Function (Oxf). 2020;1(2):zqaa024. PMID: 33201937; PMCID: PMC7662147.
- Guhr Lee TN, Cholon DM, Quinney NL, Gentzsch M, Esther CR Jr. Accumulation and persistence of ivacaftor in airway epithelia with prolonged treatment. J Cyst Fibros. 2020 Sep;19(5):746-751. PMID: 32536510; PMCID: PMC7492410.
- McCravy MS, Quinney NL, Cholon DM, Boyles SE, Jensen TJ, Aleksandrov AA, Donaldson SH, Noone PG, Gentzsch M. Personalised medicine for non-classic cystic fibrosis resulting from rare CFTR mutations. Eur Respir J. 2020 Jul 30;56(1):2000062. PMID: 32265312; PMCID: PMC7406131.
- Armirotti A, Tomati V, Matthes E, Veit G, Cholon DM, Phuan PW, Braccia C, Guidone D, Gentzsch M, Lukacs GL, Verkman AS, Galietta LJV, Hanrahan JW, Pedemonte N. Bioactive Thymosin Alpha-1 Does Not Influence F508del-CFTR Maturation and Activity. Sci Rep. 2019 Jul 16;9(1):10310. PMID: 31311979; PMCID: PMC6635361.
- Clancy JP, Cotton CU, Donaldson SH, Solomon GM, VanDevanter DR, Boyle MP, Gentzsch M, Nick JA, Illek B, Wallenburg JC, Sorscher EJ, Amaral MD, Beekman JM, Naren AP, Bridges RJ, Thomas PJ, Cutting G, Rowe S, Durmowicz AG, Mense M, Boeck KD, Skach W, Penland C, Joseloff E, Bihler H, Mahoney J, Borowitz D, Tuggle KL. CFTR modulator theratyping: Current status, gaps and future directions. J Cyst Fibros. 2019 Jan;18(1):22-34. PMID: 29934203; PMCID: PMC6301143.
- Gentzsch M, Cholon DM, Quinney NL, Boyles SE, Martino MEB, Ribeiro CMP. The cystic fibrosis airway milieu enhances rescue of F508del in a pre-clinical model. Eur Respir J. 2018 Dec 20;52(6):1801133. PMID: 30287473; PMCID: PMC6482470.
- Moorefield EC, Blue RE, Quinney NL, Gentzsch M, Ding S. Generation of renewable mouse intestinal epithelial cell monolayers and organoids for functional analyses. BMC Cell Biol. 2018 Aug 15;19(1):15. PMID: 30111276; PMCID: PMC6094565.
- Kota P, Gentzsch M, Dang Y, Boucher R, Stutts M. The N-terminus of alpha-ENaC mediates ENaC cleavage and activation by furin. J Gen Physiol. 2018 Aug 6;150(8):1179-1187. PMID: 29980634; PMCID: PMC6080898.
- Gentzsch M, Mall MA. Ion channel modulators in cystic fibrosis. Chest. 2018 Aug;154(2):383-393. PMID: 29750923; PMCID: PMC6113631. Review.
- Cholon DM, Gentzsch M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J Cyst Fibros. 2018 Mar;17(2S):S52-S60. PMID: 28986017; PMCID: PMC5828944.
- Tomati V, Caci E, Ferrera L, Pesce E, Sondo E, Cholon DM, Quinney NL, Boyles SE, Armirotti A, Ravazzolo R, Galietta LJ, Gentzsch M, Pedemonte N. Thymosin α-1 does not correct F508del-CFTR in cystic fibrosis airway epithelia. JCI Insight. 2018 Feb 8;3(3):e98699. PMID: 29415893; PMCID: PMC5821210.
- Guimbellot JS, Leach JM, Chaudhry IG, Quinney NL, Boyles SE, Chua M, Aban I, Jaspers I, Gentzsch M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight. 2017 Nov 16;2(22):e95734. PMID: 29202459; PMCID: PMC5752372.
- Gentzsch M, Boyles SE, Cheluvaraju C, Chaudhry IG, Quinney NL, Cho C, Dang H, Liu X, Schlegel R, Randell SH. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol. 2017 May;56(5):568-574. PMID: 27983869; PMCID: PMC5449492.
- Gentzsch M, Ren HY, Houck SA, Quinney NL, Cholon DM, Sopha P, Chaudhry IG, Das J, Dokholyan NV, Randell SH, Cyr DM. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol. 2016 Sep 1;311(3):L550-9. PMID: 27402691; PMCID: PMC5142211.
- Watson MJ, Lee SL, Marklew AJ, Gilmore RC, Gentzsch M, Sassano MF, Gray MA, Tarran R. The cystic fibrosis transmembrane conductance regulator (CFTR) uses its C-terminus to regulate the A2B adenosine receptor. Sci Rep. 2016 Jun 9;6:27390. PMID: 27278076; PMCID: PMC4899698.
- Cholon DM, Esther CR Jr, Gentzsch M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Rev Precis Med Drug Dev. 2016;1(3):235-243. PMID: 27482545; PMCID: PMC4963025.
- Kota P, Buchner G, Chakraborty H, Dang YL, He H, Garcia GJM, Kubelka J, Gentzsch M, Stutts MJ, Dokholyan NV. The N-terminal domain allosterically regulates cleavage and activation of the epithelial sodium channel. J Biol Chem. 2014 Aug 15;289(33):23029-23042. PMID: 24973914; PMCID: PMC4132802.
- Cholon DM, Quinney NL, Fulcher ML, Esther CR, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of ∆F508 CFTR in cystic fibrosis. Sci Transl Med. 2014 Jul 23;6(246):246ra96. PMID: 25101886; PMCID: PMC4272825.
- Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV, Riordan JR. Allosteric modulation balances thermodynamic stability and restores function of ∆F508 CFTR. J Mol Biol. 2012 May 25;419(1-2):41-60. PMID: 22406676; PMCID: PMC3891843.
- Kota P, García-Caballero A, Dang H, Gentzsch M, Stutts MJ, Dokholyan NV. Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry. 2012 Apr 24;51(16):3460-9. PMID: 22471557; PMCID: PMC3404201.
- Clunes LA, Davies CM, Coakley RD, Aleksandrov AA, Henderson AG, Zeman KL, Worthington EN, Gentzsch M, Kreda SM, Cholon D, Bennett WD, Riordan JR, Boucher RC, Tarran R. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012 Feb;26(2):533-45. PMID: 21990373; PMCID: PMC3290447.
- Kreda SM, Gentzsch M. Imaging CFTR protein localization in cultured cells and tissues. In Cystic fibrosis diagnosis and protocols. Methods Mol Biol. 2011;742:15-33. PMID: 21547724.
- Johnson JS, Gentzsch M, Zhang L, Ribeiro CM, Kantor B, Kafri T, Pickles RJ, Samulski RJ. AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis. PLoS Pathog. 2011 May;7(5):e1002053. PMID: 21625534; PMCID: PMC3098238.
- Hegedus T, Gentzsch M, Riordan JR. CFTR: Understanding the cause and influencing the outcome of a major genetic disease. In: The ABC transporters of human physiology and disease. Chapter 10. Linton K, Holland B (eds). World Scientific Publishing, 2011. pp. 269-309.
- Gentzsch M, Dang H, Dang Y, Garcia-Caballero A, Suchindran H, Boucher RC, Stutts MJ. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J Biol Chem. 2010 Oct 15;285(42):32227-32. PMID: 20709758; PMCID: PMC2952223. “Paper of the Week”
- Aleksandrov AA, Kota P, Aleksandrov L, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, Riordan JR. Regulatory insertion removal restores maturation, stability and function of ∆F508 CFTR. J Mol Biol. 2010 Aug 13;401(2):194-210. PMID: 20561529; PMCID: PMC4361937.
- Cholon DM, O’Neal WK, Randell SH, Riordan JR, Gentzsch M. Apical stability and endocytic trafficking of wild-type and mutant CFTR in primary cultures of human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2010 Mar;298(3):L304-14. PMID: 20008117; PMCID: PMC2838667.
- Hutt DM, Herman, DM, Rodrigues, APC, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Richarson J, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyonad T, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, Balch WE. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010 Jan;6(1):25-33. PMID: 19966789; PMCID: PMC2901172.
- Young A, Gentzsch M, Jia Y, Abban CY, Meneses PI, Bridges RJ, Bradbury NA. Dynasore inhibits removal of wild-type and ∆F508 CFTR from the plasma membrane. Biochem J. 2009 Jul 15;421(3):377-85. PMID: 19442237.
- Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, Saavedra MT, Salmon P, Randell SH. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol. 2009 Jan;296(1):L82-91. PMID: 18978040; PMCID: PMC2636952.
- Chang XB, Mengos A, Hou YX, Cui L, Jensen TJ, Aleskandrov A, Riordan JR, Gentzsch M. Role of N‑linked oligosaccharides in the biosynthetic processing of CFTR. J Cell Sci. 2008 Sep 1;121(Pt 17):2814-23. PMID: 18682497; PMCID: PMC2677381.
- Gentzsch M, Choudhury A, Chang X-B, Pagano R, Riordan JR. Misassembled mutant ∆F508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J Cell Sci. 2007 Feb 1;120(Pt 3):447-55. PMID: 17213331.
- Thelin, WR, Chen, Y, Gentzsch M, Kreda S, Sallee J, Scarlett C, Borchers C, Jacobson, K, Stutts KJ, Milgram S. A direct interaction with filamins modulates the stability and plasma membrane expression of CFTR. J Clin Invest. 2007 Feb;117(2):364-74. PMID: 17235394; PMCID: PMC1765518.
- Cui L, Aleksandrov L, Chang X-B, Hou YX, He L, Hegedus T, Gentzsch M, Aleksandrov A, Balch WE, Riordan JR. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007 Jan 26;365(4):981-94. PMID: 17113596.
- Hegedus T, Aleksandrov A, Cui L, Gentzsch M, Chang X-B, Riordan JR. ∆F508 CFTR with two altered RXR motifs escapes from ER quality control but its channel activity is thermally sensitive. Biochim Biophys Acta. 2006 May;1758(5):565-72. PMID: 16624253.
- Grubb BR, Gabriel SE, Mengos A, Gentzsch M, Randell SH, Van Heeckeren AM, Knowles MR, Drumm ML, Riordan JR, Boucher RC. SERCA pump inhibitors do not correct biosynthetic arrest of DF508 CFTR in cystic fibrosis. Am J Respir Cell Mol Biol. 2006 Mar;34(3):355-63. PMID: 16284361; PMCID: PMC2644200.
- Gentzsch M, Chang X-B, Cui L, Wu Y, Ozols VV, Choudhury AK, Pagano RE, Riordan JR. Endocytic trafficking routes of wild-type and DF508 CFTR. Mol Biol Cell. 2004 Jun;15(6):2684-96. PMID: 15075371; PMCID: PMC420093.
- Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem. 2003 Feb 21;278(8):6440-9. PMID: 12471024.
- Gentzsch M, Aleksandrov A, Aleksandrov L, Riordan JR. Functional analysis of the C‑terminal boundary of the second nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator and structural implications. Biochem J. 2002 Sep 1;366(Pt 2):541-8. PMID: 12020354; PMCID: PMC1222794.
- Gentzsch M, Riordan JR. Localization of sequences within the C-terminal domain of CFTR which impact maturation and stability. J Biol Chem. 2001 Jan 12;276(2):1291-8. PMID: 11022033.
Complete Bibliography:
http://www.ncbi.nlm.nih.gov/pubmed?term=Gentzsch%20M%5BAuthor%5D&cmd=DetailsSearch
