Marsico Lung Institute Accomplishments
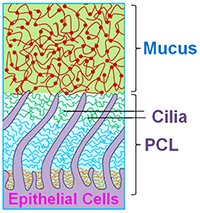
Since the inception of CF research, there has been a debate over pathogenesis of CF airways disease. Over the past decade, we have evolved the hypothesis that the CFTR-mediated ion transport defects produce a low salt and volume condition of the airway surface, which was initially termed the “low volume hypothesis.” Whereas this description likely adequately defines CF lung disease at its inception, i.e., in infants and/or areas into which CF disease is spreading within the CF lung, it has not been a satisfying all-inclusive definition because in many parts of the CF lung, there is an excess of salt and water, i.e., likely in the form of “pus.” In parallel with these concerns, the establishment of the Virtual Lung Group highlighted the inadequacies of the then current model of mucociliary clearance that posited that a slab of mobile mucins contained in the mucus layer was moving over a periciliary “liquid” layer. Both theoretical considerations (e.g., thermodynamic considerations suggesting that mucins should be moving from an area of high concentration in the mucus layer to low concentration in the PCL) and experimental observations (addition of liquids to cell cultures suggested that all the added liquid was taken up by the mucus layer and not the PCL), led to a reformulation of the nature of the PCL. In brief, as shown in Fig. 1, we now envision the PCL as a grafted polyanionic brush that has a density, and hence osmotic pressure, that exceeds that of the overlying mucus layer. This formulation suggests that in health, the osmotic pressure of the PCL should exceed that of the mucus layer so that the PCL remains a well-hydrated lubricant layer.
In CF, a depletion of salt and water on airway surfaces (“dehydration”) initially produces a selective loss of liquid from the mucus layer (note, the concentration of the PCL tethered mucins is relatively fixed, by nature of their being tethered to the ciliary/microvilli/cell surface membranes). When dehydration of the mucus layer is sufficient that the concentration of mucins in the mucus layer exceeds that of the PCL, and hence the osmotic pressure of the mucus layer exceeds that of the PCL, water is removed from the PCL, the PCL collapses, and mucus-PCL adhesion occurs. Hence, the formal definition of “dehydration of” CF airway surfaces therefore becomes the concentration of mucins in the mucus layer that produces an osmotic pressure that exceeds that of the PCL. Note, mucins in overlap conditions in solution exhibit a relationship between mucin concentration and osmotic pressure (as indexed by the distance between the nearest mucin monomers, i.e., the correlation length (ξ) that is a power function, i.e., Π mucus layer = Kt / ξ3). We believe this new formulation adequately describes the thermodynamic relationships between mucins in the two layers and water on airway surfaces, generates novel biomarkers for tracking the severity of CF lung disease (e.g., mucin concentrations), and suggests therapeutically that a goal is to re-dilute mucins on the airway surface in CF patients, i.e., rehydrate airway surfaces.
Progress in Understanding CFTR Biogenesis
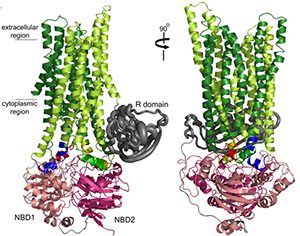
Tremendous progress at UNC has been made by the Riordan and Cyr labs in understanding the biogenesis of CFTR in health and how it fails as a consequence of mutations, e.g., DF508. An important collaboration was established upon the arrival of Dr. Riordan at UNC with the Structural Modeling Group of Dr. Nikolai Dokholyan. Based on the structure of the yeast ABC protein Sav1866, the Riordan/Dokholyan lab published the most complete model of CFTR structure in 2008 (Fig. 2). This model emphasizes the relationships in CFTR biogenesis between region of NBF1 and intracellular loop 4 that are defective in many mutant CFTRs, including D508. This model has established a paradigm for targets of small molecular “correctors” that might logically be targeted at restoring interactions between these two domains to promote the folding of mutant CFTRs. Other major advances have been made by Drs. Riordan and Cyr labs, including identification of the RI domain and its relationships to NBF1 folding, the identification of novel chaperone-CFTR interactions, and measurement of accurate half-times of both wild-type and mutant CFTR in the apical membrane of well-differentiated airway epithelial cells.
Role of ENaC and “Alternative” Cl- Channels in Relationship to CFTR Function in Health and Disease
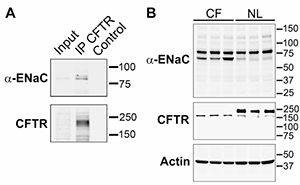
It has long been debated whether or not there is a relatively cell type-specific, i.e., airway and colon, regulatory relationship between CFTR and ENaC. Gratifyingly, insights into the molecular basis of this interaction are now emerging. Previous data from patch clamp studies and ion-selective microelectrode studies had suggested that the absolute ENaC conductance in the apical membrane of CF airways was ~3-fold higher than normal, and this reflected an increase in channel activation (increased Po). The emerging knowledge of the requirement of endoproteolysis in the activation of ENaC, i.e., increasing its Po, led to novel avenues to approach CFTR-ENaC interactions. These studies were first spurred by the data of Tarran et al. in 2006 that reported that normal airway cells exhibited a reduction in amiloride-sensitive PD as an added volume of liquid was absorbed to produce a homeostatic level of ASL on airway surfaces (~7 µm), and the amiloride-sensitive PD could be raised back to initial levels by the addition of trypsin (3). In contrast, CF airway epithelia exhibited a large amiloride-sensitive PD immediately after volume addition, which persisted despite the fact that volume was excessively absorbed – and the PD was not stimulated by trypsin addition. These findings led to the speculation that perhaps wild-type CFTR could “shield” ENaC from fully activating proteolysis – and would confer regulation by proteolysis in response to volume status. In contrast, the speculation was that in CF, ENaC may be exposed to unrestrained proteolysis and activation (increased Po) independent of volume status. Data from oocytes and human cultured cells suggest that, indeed, this hypothesis may be tenable Gentzsch et al. (J Biol Chem, 2010; 285(42):32227-32). Specifically, we observe systematically increased levels of endoproteolysis of both α and γENaC in CF cells as compared to wild-type cells. The reconstitution studies in oocytes suggest that CFTR can protect ENaC from endoproteolytic activation by specific airway epithelial cell surface proteinases that have been identified as part of the channel-activating protease family, e.g., matriptase. While still early, these studies have provided a seeming breakthrough into understanding this important relationship. For this reason, we have proposed a Pilot & Feasibility project to explore this relationship more fully by in vitro tests, and based on the reported data from Vertex770 in G551D patients that the Vertex770 effect is larger on Na+ transport than Cl– transport, we have proposed to take this observation into the clinic to develop a novel biochemical marker for restoration of CFTR regulation of Na+ transport. In an original description of the CFTR/mouse, we speculated that an “alternative” Cl– channel was protecting the CF mouse lung from the genetic deletion of the CFTR gene. In the past cycle, the extremely important work of three groups identifying a candidate gene for the “alternative” Ca2+-activated Cl- channel, i.e., TME16A, was published. We capitalized on these observations to collaborate with Jason Rock, from Brigid Hogan’s laboratory at Duke University, to test the importance of TMEM16A in the Cl– secretory capacity of murine airways, and the relationship between this capacity and mucus hydration/clearance in a variety of TMEM16A mouse models. We recently published that the complete null TMEM16A mouse exhibited a neonatal lethal phenotype that we hypothesize reflected a combination of dehydrated tracheal mucus and a hypercompliant, collapsible trachea that produced asphyxia. Thus, these data both argue strongly for the importance of the “alternative” Cl– channel in mouse airways physiologically and establish TMEM16A as an important pharmacologic target for drug therapy.
The Role of CF Airway Intraluminal Hypoxia and the CF Airway Microbiome
Approximately eight years ago, we published the observation that the CF airway lumen was frankly hypoxic in areas of mucus accumulation. Initial studies that followed that observation focused on the biology of Pseudomonas in anaerobic environments. Over the past three years, in collaboration with Drs. Michael Tunney and Stuart Elborn in Ireland, Matt Wolfgang and colleagues have added 16S T-RFLP technologies and more recently, 454 pyrosequencing technologies to sophisticated anaerobic culture techniques to define the complete CF airway microbiome.
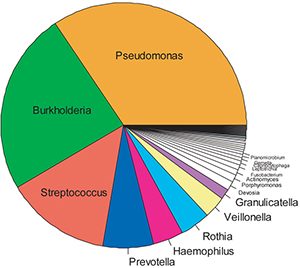
A representative “pie chart” of the spectrum of bacteria in the CF lung of the average “Irish 18-year-old CF patient” is shown in Fig. 4 below. Note the diversity of the taxa/phyla that appear to infect the CF lung, and note the large number of obligate anaerobes that occupy this niche. Thus, these studies confirm the observation that the CF airway lumen is indeed characterized by frankly anaerobic niches. They have also led to large-scale trials designed to identify the acquisition of anaerobes with respect to the other members of the community, to identify their role in CF airways pathogenesis (particularly bronchiectasis), and to develop pilot data for critical therapeutic clinical trials designed to ask whether these bacteria can and should be eradicated to improve the health of the CF lung.
16S-454 pyrosequencing. N=50 CF sputum samples.
Modifier Genes
It is clear that CF investigators have not been able to predict the outcome of either CF lung disease or CF liver disease based on CFTR genotype. Therefore, Dr. Knowles and colleagues have set about conducting systematic studies designed to identify modifier genes in both organs. With respect to the lung, initial studies of candidate genes suggested there was a relationship between polymorphisms in the TGFB1 gene and the severity of lung disease. Parallel studies in the liver revealed there were associations between mutations in the α1-antitrypsin gene and the severity of CF liver disease. More recently, a large international Consortium, in the U.S. and Canada, has focused on genome-wide association studies (GWAS) in an attempt to identify novel genes that link to CF lung disease.
In addition, Drs. Knowles and O’Neal have performed association studies of gene expression from lymphoblastoid cell lines from 754 Phe508del homozygotes to identify new modifier genes and pathways (O’Neal et al., AJHG, 2015). LPARG, a G-protein coupled receptor, associated with severity of CF lung disease (q-value = 0.0006). Pathways associated with lung disease severity were annotated in 3 broad categories: 1) endomembrane function, containing Phe508 processing genes, providing evidence of the importance of Phe508del processing to explain lung phenotype variation; 2) HLA genes, extending previous GWAS findings in the HLA region; and 3) endoplasmic reticulum stress response genes. Expression pathways were concordant for endosome and HLA pathways identified from GWAS associations in 1,978 CF patients. Pathways associated with age-of-onset of persistent P. aeruginosa were enriched for HLA Class II genes.