Deconvolution
Contents
Title: Deconvolution of Tumor Genomic Data
Introduction
Tumor samples may be a mixture of normal cell contaminations and/or different cancer subtypes. The transcriptome profiles obtained from mixed tumor samples requires a denoising process for reliable downstream analysis. Our goal is to build up powerful statistical tools to accurately segregate tumor signals from the observed genomic profile, hence facilate our understanding of tumorigenesis. This is a joint work with Dr. Wenyi Wang.
Questions
1. What is the proportion of pure tumor cells in a mixed tumor sample? (Tumor Purity Estimation)
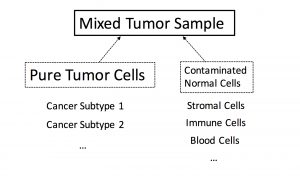
2. For a single gene, given its observed expression level from a mixed sample, how much of the amount is from pure tumor cells? (Deconvolution for Tumor Signal)
Methods
DeMix is a statistical tool developed in Wang’s lab, which assesses tumor-specific proportions and reconstitutes individual gene expressions in mixed tumor samples. A key assumption behind the model assumes the observed expression level Yig for gene g in mixed tumor samples i is the sum of gene expression level of pure tumor tissue and of the surrounding normal cells. Since the deconvolution results obtained from this model refer substantially to the distribution of genes in normal samples, it is critical to have a quality control step for these samples. We developed a pre-screening procedure to ensure there is none sample swapping issue, i.e. tumor samples mistakenly labelled as normal samples shown as below. Techniques such as the Rank Based Non-parametric Test, Principle Component Analysis, Hierarchical Clustering were employed.
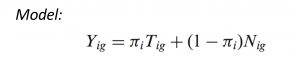

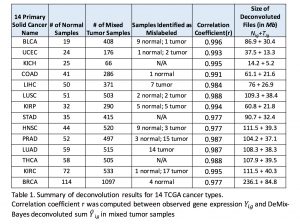
Findings
We have applied DeMix-Bayes (an advanced DeMix version under testing) to 14 primary solid cancer types at The Cancer Genome Atlas(TCGA). The denoised RNA-seq Level 3 tumor sample transcriptome profiles are available upon request.
References
Ahn, J., Yuan, Y., Parmigiani, G., Suraokar, M. B., Diao, L., Wistuba, I. I., Wang, W. (2013). DeMix: Deconvolution for mixed cancer transcriptomes using raw measured data. Bioinformatics, 29(15), 1865-1871. [PDF]