Genetic Analysis
Contents
Genetic driver factors of Concurrent cytotoxic chemotherapy and radiotherapy response for Head-Neck Squamous Cell Carcinoma
Introduction
Concurrent cytotoxic chemotherapy and radiotherapy (CXRT) forms the backbone of treatment for squamous cell carcinomas of the head and neck (HNSCC). Despite years of basic, clinical, and translational research this approach has not changed substantially since its adoption close to two decades ago. Further understanding of CXRT response and resistance is critical to improving treatment outcome and identifying novel combination therapies.
This work will focus on head and neck squamous cell carcinoma; however our approach will be applicable to other tumor types. In this work Mutation, copy number, and gene expression data from TCGA HNSCC will be analyzed in the context of signaling networks and communities to identify genomic events that alter network structure. These candidate drivers and therapeutic targets will be prioritized in the context of CTEP target genes. Additionally, genomic events will be analyzed for associations with outcome and treatment response individually and in signaling networks to identify candidate biomarkers and therapeutic targets. These drivers will then validated in our collaborator’s lab via both in vivo and in vitro experiments.
Questions
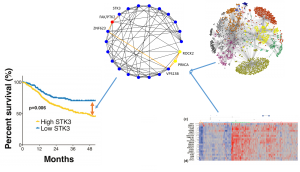
Identify driver genetic factor for CXRT response for HNSCC patients.
Methods
We proposed a method that starts with discover communities from a protein-protein-interaction network. Based on the community, we identify the key mutation/factor in both the community enrichment and structure aberration.
To assess the enrichment, we obtained a handful none malignant solid tissue samples, and tested overall gene expression difference between the non-malignant samples and the malignant samples. To acquire more statistical power, we have designed a clustering method to group the non-malignant solid samples from other organ sites that are similar to the HNSCC samples.
Findings
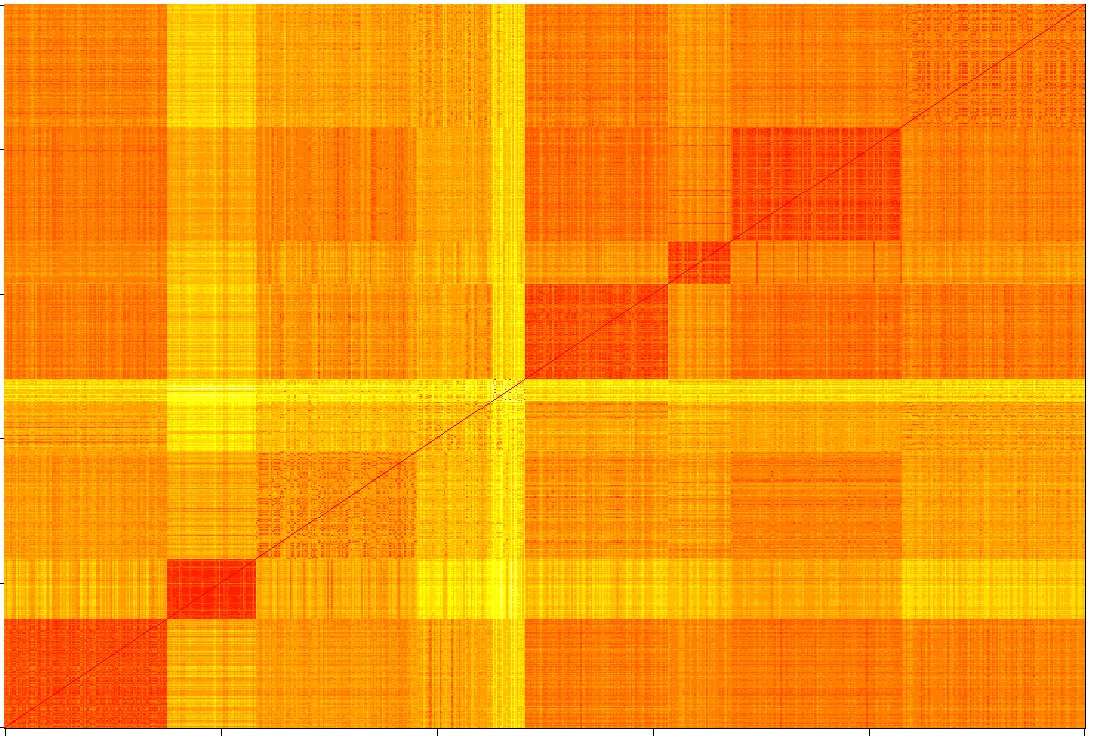
The network obtained from the gene expression of the HNSCC patients shows that there are indeed some new connections appeared in the HNSCC patients comparing to the general population. Some of the new connection after futher analysis and validation may be identified as HNSCC specific. We are able to find 109 samples that are similar to the majority of HNSCC samples, that is an increment of 150% in sample size. In the course of exploring clustering non-malignant samples, we have noticed there is heterogeneity within all the HNSCC samples. And it is likely a contamination from other cells besides the squamous cell.


References
Jin, Jiashun. Fast community detection by SCORE. Ann. Statist. 43 (2015), no. 1, 57–89. doi:10.1214/14-AOS1265.
Jerome Friedman, Trevor Hastie and Robert Tibshirani (2007). Sparse inverse covariance estimation with the lasso. Biostatistics 2007.
Meinshausen, N. and Buhlmann, P.(2006) High dimensional graphs and variable selection with the lasso. Annals of Statistics,34, p1436-1462.