Research Interests
Sickle Cell Disease
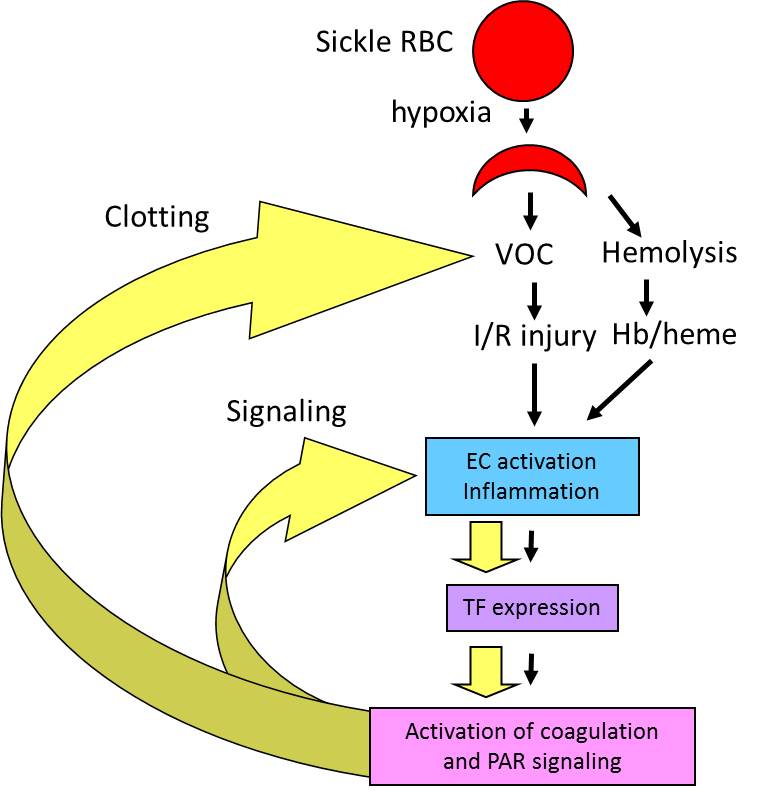 Sickle cell disease (SCD) is caused by a single nucleotide mutation that replaces glutamic acid with valine at the 6th position of the b-globin gene. Under hypoxic conditions, this single mutation leads to polymerization of hemoglobin tetramers resulting in the formation of sickled red blood cells that are less flexible, prone to hemolysis and adhere to the endothelium. This primary event results in obstruction of the microvasculature, leading to ischemic injury in multiple organs. SCD is also associated with activation of coagulation cascade. We are investigating the contribution of coagulation to the pathophysiology of SCD. We are testing the hypothesis that vaso-occlusion and hemolysis-dependent endothelial cell (EC) activation and inflammation lead to the increased expression of tissue factor (TF) and subsequent activation of coagulation. We propose that activation of coagulation creates a positive feed-back loop that further enhances vaso-occlusion, EC activation and inflammation via activation of various protease activated receptors (PARs). We believe that targeting coagulation will not only have anti-thrombotic effects but may also reduce inflammation and EC dysfunction.
Sickle cell disease (SCD) is caused by a single nucleotide mutation that replaces glutamic acid with valine at the 6th position of the b-globin gene. Under hypoxic conditions, this single mutation leads to polymerization of hemoglobin tetramers resulting in the formation of sickled red blood cells that are less flexible, prone to hemolysis and adhere to the endothelium. This primary event results in obstruction of the microvasculature, leading to ischemic injury in multiple organs. SCD is also associated with activation of coagulation cascade. We are investigating the contribution of coagulation to the pathophysiology of SCD. We are testing the hypothesis that vaso-occlusion and hemolysis-dependent endothelial cell (EC) activation and inflammation lead to the increased expression of tissue factor (TF) and subsequent activation of coagulation. We propose that activation of coagulation creates a positive feed-back loop that further enhances vaso-occlusion, EC activation and inflammation via activation of various protease activated receptors (PARs). We believe that targeting coagulation will not only have anti-thrombotic effects but may also reduce inflammation and EC dysfunction.
Heart I/R injury, myocardial infarction and heart remodeling.
Myocardial infarction induced by ischemia/reperfusion (I/R) injury is a major clinical problem. During I/R injury, damage to the endothelial barrier allows a leakage of coagulation factors into the myocardium. Tissue factor (TF), the primary activator of the coagulation cascade, is constitutively expressed in the heart. Activation of coagulation cascade by TF has been shown to play a role in myocardial infarction after I/R injury. However, the role TF:FVIIa complex signaling has not been investigated.
The TF:FVIIa complex can activate cells by cleavage of protease activated receptor-2 (PAR-2). Both TF and PAR-2 are expressed on cardiomyocytes as well as neutrophils, which infiltrate into the myocardium after I/R injury. We have demonstrated that PAR-2 deficiency results in significant reduction of infarct size, heart remodeling and heart dysfunction after I/R injury. We are trying to determine if TF:FVIIa-dependent activation of PAR-2 contributes to myocardial infarction and heart remodeling. Using unique set of genetic and pharmacologic tools we are trying to distinguish signaling and coagulation properties of TF:FVIIa complex. Understanding the role of TF:FVIIa-dependent activation of PAR-2 in injured heart may lead to the development of new therapies to reduce myocardial infarction and heart failure.
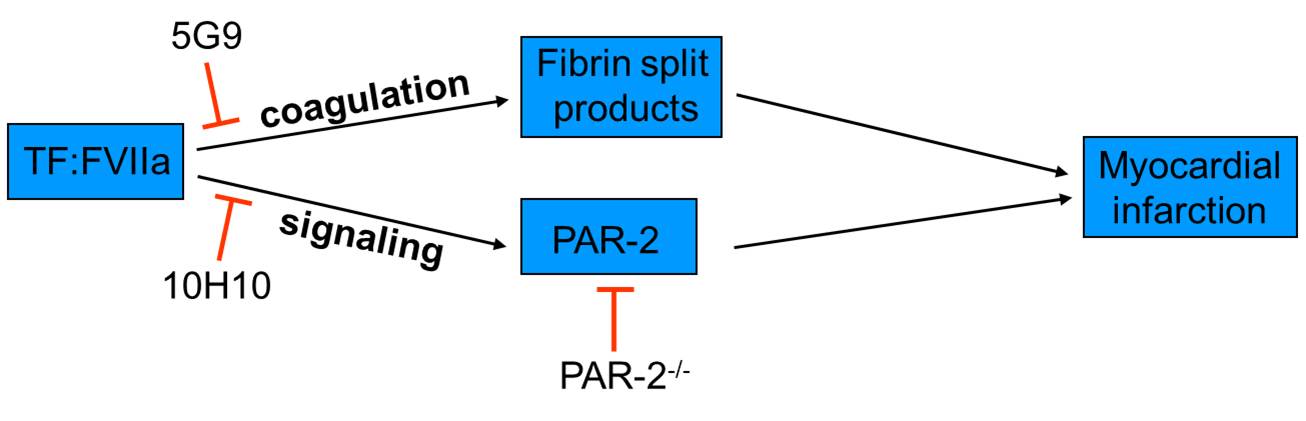
(i) Activation of the coagulation cascade results in thrombin generation and fibrin deposition. Fibrin degradation products facilitate neutrophil transmigration into the myocardium via a VE-cadherin-dependent mechanism; (ii) The TF:VIIa complex contributes to MI by activating PAR-2. 5G9 blocks binding of FX to TF:FVIIa complex without interfering with TF:FVIIa-dependent activation of PAR-2. 10H10 blocks TF:FVIIa-dependent activation of PAR-2 with no effect on coagulation.
Neuroinflammation
Coming soon