Research Interests
Key words: evolution, mouse genetics, epigenetics, female meiosis, chromosome segregation, meiotic drive.
“Nothing in Biology makes sense except in the light of evolution” T. Dobzhansky (1973).
This quote summarizes my research philosophy and the unifying theme of the work conducted in my laboratory. Although most biomedical researchers probably agree with Dobzhansky famous statement, only a minority of them uses such principle as a guidance to formulate their research questions, to design their experiments and to interpret their results. I believe that this represents a severe shortcoming and it is profoundly dysfunctional situation in the post-genome era.
The large investments of the past decade in genome projects and more recently in genetic diversity (hapmap) projects are now bearing their first fruits. Genome wide association studies in humans and comparative genomics studies are two areas in which extraordinary advances are currently been made. The laboratory mouse has been the most popular, and arguably the successful, mammalian model to investigate human physiology and disease, while its feral counterpart is an exceptional model to address evolutionary questions. However, within the mouse community the paradigm shift required to switch from single genes to systems genetics and the full incorporation of new genetic/genomic/evolutionary data and tools to investigate every biological process has not yet taken broad hold (Churchill et al. Nature Genetics 2004). Given the privileged position of the mouse among mammalian systems it is difficult to overstate the potential for such paradigm shift to revolutionize biomedical research. My contribution to this paradigm shift is to incorporate state-of-the-art evolutionary approaches and to include evolutionary considerations to propose, design and interpret all aspects of biomedical research in the mouse. I am interested in probing long, but normally poorly tested assumptions that underlie the evolution of the mammalian genome and its implication for human disease. In the following paragraphs I provide two short examples recently published.
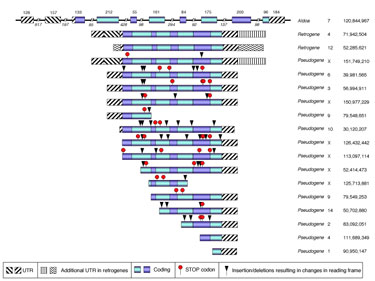 My laboratory collaborated with Dr. Deborah O’Brien (uNC) in the identification and functional characterization two new mouse genes encoding for isoforms of a glycolytic enzyme. The study was motivated by our collaborator long standing interest in the critical role that this pathway plays in male fertility mediated primarily by its requirement for sperm motility. What makes this study compelling for us is that by using a combination of bioinformatics, evolutionary and comparative genomics tools we demonstrated that these isoforms are encoded by novel retrogenes that are members of a large family of related sequences that arose by multiple retrotransposition events that have occurred recurrently in all mammalian lineages including humans (Vemuganti et al. Developmental Biology 2007). We also identified a novel N-terminal extension of the protein and predicted the tissue and developmental stage in which those genes are expressed. These predictions have all been confirmed by RT-PCR. This is not an idiosyncrasy of these particular genes. We have shown that all the enzymes of the glycolityc pathway are also characterized by recurrent retrotransposition events, emergence of retrogenes with male germline restricted expression patterns and dramatic sequence divergence possibly related to positive selection for sperm functional characteristics and speciation events (D. O’Brien and F. Pardo-Manuel de Villena; unpublished results). Dr. O’Brien will focus on the physiological, cellular and biochemical relevance of these genes in male fertility. I will explore the genetic and genomic features associated with high rates of retrotransposition and with the emergence and maintenance of functional retrogenes (preliminary results suggest the involvement of epigenetic signals and an enrichment for ultraconserved elements).
My laboratory collaborated with Dr. Deborah O’Brien (uNC) in the identification and functional characterization two new mouse genes encoding for isoforms of a glycolytic enzyme. The study was motivated by our collaborator long standing interest in the critical role that this pathway plays in male fertility mediated primarily by its requirement for sperm motility. What makes this study compelling for us is that by using a combination of bioinformatics, evolutionary and comparative genomics tools we demonstrated that these isoforms are encoded by novel retrogenes that are members of a large family of related sequences that arose by multiple retrotransposition events that have occurred recurrently in all mammalian lineages including humans (Vemuganti et al. Developmental Biology 2007). We also identified a novel N-terminal extension of the protein and predicted the tissue and developmental stage in which those genes are expressed. These predictions have all been confirmed by RT-PCR. This is not an idiosyncrasy of these particular genes. We have shown that all the enzymes of the glycolityc pathway are also characterized by recurrent retrotransposition events, emergence of retrogenes with male germline restricted expression patterns and dramatic sequence divergence possibly related to positive selection for sperm functional characteristics and speciation events (D. O’Brien and F. Pardo-Manuel de Villena; unpublished results). Dr. O’Brien will focus on the physiological, cellular and biochemical relevance of these genes in male fertility. I will explore the genetic and genomic features associated with high rates of retrotransposition and with the emergence and maintenance of functional retrogenes (preliminary results suggest the involvement of epigenetic signals and an enrichment for ultraconserved elements).
 In the second example our proximate goal was to generate a genotyping array that could be used to genotype all existing mouse strains and resources. Existing arrays have low marker density, there are systemic problems associated with ascertainment bias and they are either not commercially available or their cost is prohibitive.
In the second example our proximate goal was to generate a genotyping array that could be used to genotype all existing mouse strains and resources. Existing arrays have low marker density, there are systemic problems associated with ascertainment bias and they are either not commercially available or their cost is prohibitive. 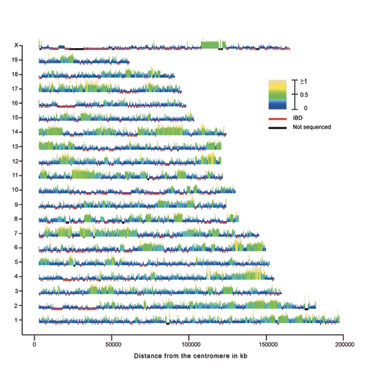 In collaboration with Dr. Gary Churchill (Jackson Laboratory) we set out to use the 109 million mouse genotypes recently released by NIEHS to examine the ancestral subspecific origin of classical inbred strains (Yang et al. Nature Genetics 2007). Based on known history we expected to identify a mosaic of segments that could be unambiguously assigned to one of three distinct lineages: M. m. domesticus, M. m. musculus and M. m. castaneus. Our plan was to use the three wild-derived strains as a reference for each subspecies and then assign genomic segments from classical and hybrid strains to a particular subspecies based on the pattern of SNP similarity between the query strain and the reference strains. Our critical contribution was to generate the analytic methods to determine the phylogenetic origin of the variation found in each genomic region taking into account the putative presence of lineage-specific differential SNP discovery rate and introgression (contamination) in the reference strains. Our conclusions (Yang et al. Nature Genetics 2007) are in sharp contrast with the prevailing view of the mouse genome (the mosaic concept) and with a competing analysis. I believe that these differences are due to the fact that we specifically designed our analyses within a well defined phylogenetic framework and that we tested the most basic assumptions (including the possibility that mouse inbred strains may not be what they have been assumed to be for decades). In addition to provide the desired catalog of variation to include in our genotyping array, these studies have the potential to revolutionize all aspects of mouse genetics. We are now investigating the implications for complex trait analysis and QTL mapping, while we are generating a high resolution map of historical recombination in the mouse genome, and determining the frequency and length of gene conversion events and the recurrent mutations.
In collaboration with Dr. Gary Churchill (Jackson Laboratory) we set out to use the 109 million mouse genotypes recently released by NIEHS to examine the ancestral subspecific origin of classical inbred strains (Yang et al. Nature Genetics 2007). Based on known history we expected to identify a mosaic of segments that could be unambiguously assigned to one of three distinct lineages: M. m. domesticus, M. m. musculus and M. m. castaneus. Our plan was to use the three wild-derived strains as a reference for each subspecies and then assign genomic segments from classical and hybrid strains to a particular subspecies based on the pattern of SNP similarity between the query strain and the reference strains. Our critical contribution was to generate the analytic methods to determine the phylogenetic origin of the variation found in each genomic region taking into account the putative presence of lineage-specific differential SNP discovery rate and introgression (contamination) in the reference strains. Our conclusions (Yang et al. Nature Genetics 2007) are in sharp contrast with the prevailing view of the mouse genome (the mosaic concept) and with a competing analysis. I believe that these differences are due to the fact that we specifically designed our analyses within a well defined phylogenetic framework and that we tested the most basic assumptions (including the possibility that mouse inbred strains may not be what they have been assumed to be for decades). In addition to provide the desired catalog of variation to include in our genotyping array, these studies have the potential to revolutionize all aspects of mouse genetics. We are now investigating the implications for complex trait analysis and QTL mapping, while we are generating a high resolution map of historical recombination in the mouse genome, and determining the frequency and length of gene conversion events and the recurrent mutations.
These two examples are also proof of my strong belief in collaborative research. The overall goal of the research in my laboratory is to advance science in a non incremental fashion by re-examining some of the basic assumptions build-in our models (questions that in many case could not be answered a few years ago). These assumptions span the gamut from the expectation for the universal operation of Mendel’s laws to the structure, levels and origin of the genetic variation present in the most basic tool for mouse genetics, the inbred strains. I expect that this research will be instrumental in building a better mouse model for biomedical research. In the following sections I briefly summarize the ongoing research.
A) Population Genetics of Inbred Strains (Project 1, Center for Genome Dynamics, 1P50 GM076468-01)
Specific Aims: The fundamental paradigm of the work at the Center for Genome Dynamics derives from our understanding that the physical and functional organization of the genome is a consequence of its evolution, and that this organization can be deciphered by exploiting the unique evolutionary experiment inbred strains of mice provide (Yang et al. Nature Genetics 2007). Doing so require that the genomic markers (SNPs) we use for mapping and the functional allelic variation they tag arose in the same branches of the evolutionary tree, that the density of our markers approximates average gene densities, and that we can carry out the requisite genotyping in a cost effective manner. Because the SNPs described in existing databases do not meet these requirements, we have used 109 million genotypes obtained by microarray resequencing of 15 inbred strains, including representatives from each of the three major mouse subspecies to generate two sets of 25,400 phylogenetic trees. Each sets contain the tree for each consecutive 100 kb interval, and the second set is displaced 50 kb with respect to the first. In each interval we determined all the strain distribution patterns (SDPs) represented in the tree and their respective frequencies. This SDP database was then used to select 400,000 SNPs representing each one of the phylogenetic branches observed in the local trees. Computationally, each segment represents a polyallelic system in which we know the time in evolution when the alleles arose, and we have an efficient means of typing these alleles across an extensive sample of inbred strains. The resulting data will 1) provide considerably improved maps of linkage disequilibrium (LD) domains and networks, 2) allow us to investigate the evolutionary forces responsible for the assembly of the LD domains and networks, 3) improve the reliability/resolution of in silico QTL mapping, 4) identify and map historical recombination events and relate these to current maps of recombination hotspots, and 5) address several basic evolutionary questions for which the genus Mus is exceptionally well suited, primary among them being the validity of Wright’s Shifting Balance theory. Specifically, we propose to:
- Establish a collection of DNA from a comprehensive set of inbred strains.
- Identify an unbiased set of ≈400,000 SNPs representing the diversity present among four mouse subspecies, M. m. domesticus, M. m. musculus, M. m. castaneus and M. m. molossinus.
- Genotype these SNPs on 2,000 mouse strains and individual samples.
- Generate a genome-wide map of the phylogenetic origin of each genomic region in each strain.
Future Directions
This project has a natural extension into two questions of particular interest to us, the dynamics of speciation in mammals and the evolutionary dynamics of the highest order of genome organization, i.e. the karyotype. Speciation is a key process in evolution and integration of the molecular data generated in this project with the resources gathered by field biologist in hybrid zones provide an exciting opportunity to take advantage of the mouse as an evolutionary model (see section B). In addition, the genome is organized in chromosomes, complex structures with to dual functions. On one hand, the genetic information is encrypted within the DNA molecule. On the other hand, chromosomes are delivery systems that ensure the stable transmission of the genetic information to the products of each cell division. Centromeres are key loci for the later function and remain at the frontiers of biological research in the post genome era. We have recently proposed the centromeric drive theory to explain centromere/karyotype evolution (Pardo-Manuel de Villena and Sapienza Genetics 2001). The different M. musculus subspecies provide a unique experimental setting in which to test this theory and to uncover the genetic basis of variation in centromere function (see section C).
B) Role of epistatic selection during population admixture and inbreeding (R01 Interdisciplinary Consortium on the Genetics and Co-Morbidity of Stress (ICOGS), 1 U54 RR024345-01, under review)
The scope and form of epistatic selection are key questions in several evolutionary processes, including speciation and adaptation. Intrinsic reproductive isolation between new species is primarily caused by dysfunctional interactions in hybrids between alleles that evolved in separate populations. Epistatic selection will act to remove these deleterious combinations from hybrid populations. Additionally, competing mechanisms for the process of adaptation postulate different roles for epistatic selection. Selection targeting multi-locus combinations re-shuffled by genetic drift can contribute to adaptive evolution; alternatively selection might primarily target mutations with additive effects. Unfortunately, quantification of epistatic selection in nature is extremely challenging; controlled experiments in the laboratory have been much more informative on this issue. We propose to use the unique resource generated by the ICOGS to measure the form and magnitude of epistatic selection. This resource consists of hundreds of recombinant inbred mouse lines (and the ability to genotype at any given density each one of their progenitors) resulting from crosses among divergent strains that are part of the Collaborative Cross. The most relevant characteristic of the Collaborative Cross is that three of the parental strains used to derive the lines belong to a different mouse subspecies, and the remaining strains are mosaics of these subspecies (Churchill et al. Nature Genetics 2004; Roberts et al. Mammalian Genome 2007). During the derivation of the recombinant inbred (RI) lines unfavorable heterospecific combinations of alleles are brought together in the mixing generations and purged from the population in the inbreeding generations. When this occurs, gametic disequilibrium ensues, allowing us to detect the action of epistatic selection and reconstruct networks of co-adapted alleles. In addition, using the same resource we will also study the role of selfish systems, in which selection operates directly on components of the genome, independently of Darwinian fitness. We propose to complete the following specific Aims:
Specific Aim 1
Measure the form and magnitude of epistatic selection operating in the derivation of the Recombinant Inbred Lines (RI lines). Our hypothesis is that during the derivation of the RI lines of the CC, epistatic selection is driven by “Dobzhansky-Muller incompatibilities”. Such incompatibilities are due to the disruption of gene interactions between combinations of favorable alleles. Therefore, we expect to find gametic disequilibrium as the result of the preferential survival of RI lines with conspecific association of alleles (alleles arising in the same subspecies) and a deficit of RI lines with heterospecific association of alleles at these loci (alleles arising in the different subspecies).
Specific Aim 2
Identify putative selfish systems operating during the derivation of the RI lines. Meiotic drive systems are selfish genetic systems that increase the frequency of the favored alleles in a population independently of organismal fitness. Drive leads to segregation distortion signatures that are similar to those of natural selection. Crosses between divergent populations with little or no gene flow between them are predicted to be especially suited to uncover meiotic drive systems (Pardo-Manuel de Villena and Sapienza Mammalian Genome 2001). The underlying rationale is that favored alleles at driven loci are expected to reach homozygosity in a panmictic population precluding further operation of the drive until heterozygosity is re-introduced by hybridization.
Specific Aim 3
Identify loci associated with extinction in emerging RI lines. A fraction of the recombinant lines initiated in the Collaborative Cross will become extinct during inbreeding. Extinction is most likely to be due to poor reproductive performance (infertility) due to sterility in either parent, incompatibility between the combination of alleles in the parental genomes and embryonic or postnatal survival, extreme sex-ratio distortion, or poor parental care.
Specific Aim 4
Determine how epistatic selection, meiotic drive, and poor reproductive performance shape the genetic landscape in the surviving RI lines. We will classify RI lines as genetically “robust” and “delicate” based on the combination of alleles present at loci: i) under epistatic selection, ii) subject or causing meiotic drive and iii) associated with poor reproductive performance. We hypothesize that “delicate” lines will be phenotypic outliers in challenging environmental situations, such as stress, because they have carry maladaptive combination of alleles at multiple pair of loci.
C) Molecular dissection of meiotic drive in the mouse (completed support from NSF)
It is generally assumed that chromosomes segregate randomly to the products of meiosis ensuring equal representation of alleles in the gametes. We have previously reported two meiotic drive systems in the mouse one operating at the first meiotic division and the other at the second meiotic division (Pardo-Manuel de Villena et al. Genetics 2000; Pardo-Manuel de Villena and Sapienza Genetics 2001).
Meiotic drive sat the first meiotic division. Such systems are widespread in plants and animals (Pardo-Manel de Villena and Sapienza Mammalian Genome 2001) and include chromosome rearrangements in insects, chicken, mouse and humans. A common feature among them is that the Responder has been mapped to the centromere or a locus tightly linked to the centromere. The functional heterozygosity at the Responder that is required for meiotic drive is caused by the presence of an odd number of centromeres, by epigenetic differences at the centromeres or by predicted differences in centromere function of unknown origin. Within a species and a system, meiotic drive consistently selects for the same type of insensitive allele at the Responder. For example, in heterozygous female mouse carriers of Robertsonian translocations, nonrandom segregation at MI selects the normal acrocentric chromosomes independently of the particular chromosomes involved in the rearrangement. However, within the M. m. domesticus subspecies many local populations have reverted the drive polarity and select for the Robertsonian chromosome (Pardo-Manuel de Villena 2005). We will use the genotyping array developed in section A to perform genome wide association in a large collection of domesticus mice from western Europe with or without translocations to identify the locus (loci) involved in polarity reversal. Given that the direction of selection varies among species this study offers the tantalizing possibility of addressing centromere function and functional asymmetry of the meiotic spindle. The later is a general feature of meiosis and can be interpreted as differences in spindle pole “strength” (Pardo-Manuel de Villena and Sapienza Mammalian Genome 2001).
Meiotic drive at the second meiotic division. We have demonstrated transmission ratio distortion for maternal alleles at the Om locus on mouse chromosome 11. This distortion results from the preferential segregation of chromatids carrying the maternal DDK allele at Om to the ovum and reciprocal preferential segregation of chromatids with the C57BL/6 allele to the second polar body. Our studies demonstrate that the Om locus acts as the Responder in this meiotic drive system. We have also shown that whether there is random segregation or meiotic drive depends on the genotype of the fertilizing sperm. Therefore, in this meiotic drive system the Distorter is provided by the sperm. This work indicates that the sperm may influence the pattern of maternal inheritance. Ongoing experiments have mapped the Distorter and Responder to sizes amenable to molecular dissection. The experiments proposed will identify at the molecular level the Distorter in the Om meiotic drive system and examine its role in spermatogenesis, fertilization and egg activation.
Publications
Lab Members
|
Post-doctoral Fellow | |
|
Graduate Student | |
|
Graduate Student | |
|
Undergraduate Student | |
|
Technician | |
|
Technician | |
|
Technician | |
|
Technician | |
|
Technician |
Collaborative Cross Project
|
Project Manager | |
|
Technician | |
|
Technician | |
|
Technician | |
|
Technician |
Fernando Pardo-Manuel de Villena in UNC Genetics News

May 19, 2026
Science Publishes Genetic Quality Control Policy Article
A team of scientists from the UNC-CH Department of Genetics and partnering institutions, led by Dr. Fernando Pardo-Manuel de Villena (Professor and Chair, UNC Genetics), published a policy article titled “Improve genetic quality control to increase rigor and reproducibility of mouse research” in the 14 May 2026 issue of Science.

September 29, 2025
Genetics Chair Awarded Academic Investment Funds for Genetic QC Program
Dr. Fernando Pardo Manuel de Villena (Professor and Chair, Genetics) was awarded Academic Investment Funds from the School of Medicine for his application titled “Pilot program to elevate genetic quality control standards and increase rigor and reproducibility of mouse-based research”.
January 25, 2024
Dr. Fernando Pardo-Manuel de Villena Renews Service Agreement with Neogen Corporation
Fernando Pardo-Manuel de Villena (Professor and Chair, Department of Genetics) has renewed a long-standing service agreement with Neogen Corporation.

July 3, 2023
Department of Genetics Publications for June 18th – July 1st, 2023
Department of Genetics faculty, postdocs, students and collaborators published 14 papers during June 18th - July 1st 2023.

April 28, 2023
Dr. Fernando Pardo-Manuel de Villena to Deliver 42nd Annual Jim McGinnis Memorial Lecture at Duke University
Dr. Fernando Pardo-Manuel de Villena to Deliver 42nd Annual Jim McGinnis Memorial Lecture at Duke University on Thursday, May 4, 2023 at 3pm in Duke's Bryan Research Building.

March 27, 2023
Genetics Hosts Wake Tech Community College (WTCC) START Program Students
On March 24, the department hosted a group of eight students who participate in the WTCC STEM Academic and Research Training (START) Program.
January 2, 2023
Department of Genetics Publications for December 18th – 31st, 2022
Department of Genetics faculty, postdocs, students and collaborators published 10 papers during December 18th – 31st, 2022.
August 16, 2022
Department of Genetics Publications for July 31st – August 13th, 2022
Department of Genetics faculty, postdocs, students and collaborators published 7 papers during July 31st – August 13th, 2022. A multi-layer functional genomic analysis to understand noncoding genetic variation in lipids. Ramdas S, Judd J, Graham SE, Kanoni S, Wang Y, Surakka I, Wenz B, Clarke SL, Chesi A, Wells A, Bhatti KF, Vedantam S, …
August 1, 2022
Department of Genetics Publications for July 17th – 30th, 2022
Department of Genetics faculty, postdocs, students and collaborators published 11 papers during July 17th – 30th, 2022.
July 18, 2022
Department of Genetics Publications for July 3rd – 16th, 2022
Department of Genetics faculty, postdocs, students and collaborators published 12 papers during July 3rd – 16th, 2022.