Research Interests
- Molecular mechanisms of activation of NLR family signaling proteins
- Mechanisms of NLRP3-inflammasome mediated programmed necrotic cell death (pyronecrosis) and caspase-1 activation
- Role of NLRP3-inflammasome signaling in infectious disease pathogenesis
Research Synopsis
The innate immune system senses pathogens utilizing cellular signaling pathways that respond to molecular patterns common to groups of microbes, known as Microbe Associated Molecular Patterns (MAMPs). These MAMP sensing pathways are critical for organisms to respond to pathogens by triggering host defensive mechanisms that prevent overwhelming infection. However, pathogens have evolved mechanisms that allow them to avoid or exploit the host responses triggered by these signaling pathways. The study of innate immune signaling and its interface with pathogenesis of infectious diseases could potentially guide the development of novel therapies for infectious diseases that act by reducing virulence of pathogens or bolstering host resistance to invading microbes.
Our lab has three foci of research aimed at understanding how manipulation of innate immune signaling can be targeted to treat or prevent infections by two pathogens recognized as emerging global threats: Staphylococcus aureus andNeisseria gonorrhoeae1.
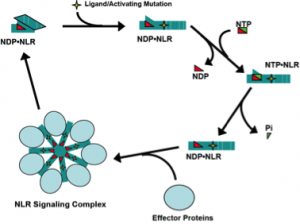
Biochemical and molecular dissection of NLR signaling pathway mechanisms.
Nucleotide-binding domain-, Leucine rich Repeat- containing proteins (also NOD-like receptors or NLRs) are a family of ATP-binding proteins involved in innate immune signaling. We study the molecular mechanisms of signaling by three NLRs: NOD2, NLRP3, and NLRC4. NOD2 and NLRC4 are required for host responses to specific bacterial molecules: peptidoglycan-derived muramyl-dipeptide and conserved protein structures from Type III Secretion Systems (T3SS). NLRP3 is activated by a diverse group of danger signals, including cytolytic toxins, purogenic receptors, and microscopic crystals. Missense mutations in all three NLR genes are associated with inherited inflammatory disorders. NOD2 signaling activates the transcription factor NF-kB and induces autophagy of intracellular pathogens. NLRP3 and NLRC4 form a cytokine-processing, proteolytic complex known as the inflammasome. Inflammasomes activate caspase-1, responsible for proteolytic maturation of IL-1b and IL-18, and initiate programmed inflammatory cell death or necrosis.
We previously demonstrated that purified, recombinant NOD2 and NLRP3 are ATPases and that NOD2 directly binds to muramyl-dipeptide. NLRC4 was subsequently shown to associate with T3SS protein-bound NAIPs (also NLR family members). Because NLRP3 is activated by many unrelated stimuli, the ligand responsible for its activation long eluded identification. We have begun to interrogate intracellular candidate ligands to determine whether there is a common cell derived ligand that is generated by these diverse NLRP3-stimulating conditions. We hypothesize that NLR ligands and autoimmune-associated mutations induce NLR signaling through effects on the ATP/ADP binding cycle (Fig. 1). Using our capacity to generate purified NLR proteins and deploy biochemical assays to measure their activation, we will determine the role of ligand binding in regulation of the ATP binding cycle and formation of signaling complexes by NOD2, NLRC4, and NLRP3. Understanding the molecular mechanisms of NLR signaling will lead to new strategies for treating autoimmune and infectious diseases in which these proteins contribute to dysregulated inflammation.
NLRP3-inflammasome activation in Staphylococcus aureus infection.
Staphylococcus aureus (Sa) infections are a growing microbial threat worldwide. Resistance to anti-staphylococcal penicillins and virulence determinants have co-emerged in circulating Sa strains, resulting in antibiotic resistant bacteria that are likely to cause severe disease. These community acquired methicillin resistant Sa (CA-MRSA) account for half of all skin infections seen in emergency rooms and can cause severe necrotizing infections with excessive mortality even in young, otherwise healthy patients. Understanding the mechanisms of CA-MRSA pathogenesis is critical to identifying adjunctive therapies to combat these infections.
Sa produces several cytolytic toxins including a-hemolysin and leukocidins. Whilea-hemolysin targets epithelial cells, endothelial cells, and immune cells, leukocidins are potently cytolytic only towards immune cells. We found that a-hemolysin, Panton Valentine Leukocidin, and LeukocidinAB all activate the NLRP3-inflammasome, inducing IL-1b secretion and initiating necrotic cell death. The convergence of these toxins on a single host signaling system suggests that activation of the NLRP3-inflammasome is advantageous to Sa. We have shown that Sa a-hemolysin activates the NLRP3-inflammasome during CA-MRSA pneumonia. Activation of NLRP3 by a-hemolysin in pneumonia is deleterious to the host rather than protective. Our findings suggest that a-hemolysin co-opts the normal protective functions of the NLRP3-inflammasome to enhance virulence of Sa. The role of a-hemolysin-mediated NLRP3-inflammasome activation in non-immune cells and the importance of leukocidin-induced NLRP3 activation in Sainfection pathogenesis remain open questions we are studying.
Host immune manipulation in Neisseria gonorrhoeae infection.
Neisseria gonorrhoeae (Ng), the most common sexually transmitted bacterial pathogen worldwide, is an exclusive human pathogen well adapted to life in the human genital tract. At the mucosal surface, Ng can cause localized inflammation. In men, this leads to copious urethral discharge. However, in ~50% of infected women, Ng is clinically silent and seemingly undistinguished from other vaginal commensal bacteria by its host. In both cases, Ng evades the development of protective immune responses, retaining the capacity to re-infect the same host.
We have found that mutant Ng, lacking hexaacylated- or phosphoethanolamine(PEA)-modified lipo-oligosaccharide (LOS), induce diminished innate immune signaling and exhibit increased sensitivity to host antimicrobial responses. These mutant Ng cause little local inflammation in a mouse model of gonorrhea, resembling asymptomatic human infection. Though infectious on their own, these mutant Ng are compromised in competitive infection with their parental strains in mice and in experimental intra-urethral infection of male human subjects. These experimental data and observations of natural Neisseria species suggest that Ng induce host inflammation to create a competitive advantage over the commensal flora. We have also reported that Nghas immune-suppressive effects on dendritic cells (DCs), which facilitate CD4+T-cell proliferation and polarization in response to pathogen-derived antigens. Our preliminary data indicates Ng sheds outer membrane vesicles (OMVs) that suppress DC-induced T-cell proliferation. We are utilizing our unique capacity to carry out human experimental infection with isogenic Ng strains to study along with murine and cell based studies to determine the gonococcal and host factors that are involved in these distinct immunopathologic components of gonococcal infection.
