FAQs
Getting Started
What is Next Generation Sequencing?
Next generation sequencing (NGS) is defined as technology allowing one to determine in a single experiment the sequence of a DNA molecule(s) with total size significantly larger than 1million base pairs (1million bp or 1Mb).
How do we get started with our sequencing project?
If you have questions, please contact htsf@med.unc.edu. A meeting with Piotr Mieczkowski, the HTSF Research Technologies Director, may also be required. We will discuss your scientific objectives so we can suggest options that will meet your data goals and conserve funds.
Are the HTSF’s services open to researchers that are not affiliated with UNC?
The HTSF is open to all researchers at UNC-CH, other academic institutions, industry and international groups.
HTSF customer service staff will assist with the submission of materials to our database system, TracSeq. The system is only accessible to UNC staff. All US researchers outside of UNC-CH will need to provide a copy of a valid purchase order before work can commence. International researchers are required to prepay for proposed work. The HTSF will provide an official invoice and electronic payment instructions for the researcher to submit to their accounting department, and the researcher will need to provide a check number and the name of the associated bank to HTSF.
The fees for service will have an F&A fee added as required by UNC Chapel Hill. HTSF does not control the percent F&A we are required to add. UNC annually determines the F&A percent.
I have an existing TracSeq account, do I need a new account for every submission?
A new account is not needed for each experiment. The submissions are differentiated by unique batch numbers, which allows for multiple submissions/experiments under the same account.
Submitting to the HTSF
How should I ship my samples?
If you are unable to physically drop off your samples to the HTSF, you are welcome to ship your samples instead. Detailed information on how to ship your raw material, libraries, or pools can be found in the HTSF Shipping and Packing Instructions document.
Can I submit any number of samples?
UNC HTSF has no minimum sample size.
How do I drop my samples off at the HTSF, and what do I need to bring with me?
When completing drop off, please bring your samples and your confirmed manifest to the HTSF. The manifest can be printed from your computer after approving your quote from TracSeq. The project coordination team will schedule a time for sample drop-off. If you are unsure where HTSF is located, see Where is the HTSF?.
Can I submit my samples in a 96-well plate?
Yes, if you are submitting more than 24 samples, we recommend using a plate instead of tubes. Plates must be submitted in column format (e.g. A1, B1, C1, … H1, then A2, B2, C2, … H2). See Submission tab for more details on plate submission.
Plates must be BioRad 96 well Skirted PCR plate, hard-shelled (Catalog number: HSP-9631). See the HTSF Approved Container Requirements.
How do I ensure that my DNA is high quality?
Please reference the HTSF DNA and RNA Sample Preparation Requirement Guide for minimum input for library preparations.
Make sure it:
- Has undergone as few freeze-thaw cycles as possible. (HMW DNA is best kept at 4°C. For procedures that require long stranded DNA, please contact the HTSF for the best extraction methods.)
- Has not been exposed to high temperatures (>65°C for 1 h can cause a detectable decrease in sequence quality).
- Has not been exposed to pH extremes (<6 or >9).
- Does not contain insoluble material and is not colored or cloudy.
- Does not contain RNA. Always RNAse treat the DNA.
- Has not been exposed to intercalating fluorescent dyes or UV radiation.
- Does not contain chelating agents (e.g., EDTA), divalent metal cations (e.g. Mg 2+), denaturants (e.g. guanidinium salts, phenol), or detergents (e.g. SDS, Triton-X100).
- Does not contain carryover contamination from the starting organism/tissue (e.g. heme, humic acid, polyphenols).
How do I ensure that my RNA is high quality?
Please reference the HTSF DNA and RNA Sample Preparation Requirement Guide for minimum input for library preparations. Additional suggestions can be found under Submissions, on the Sample Preparation Requirements for Submission page.
Make sure RNA:
- is stored at -80
- Has undergone as few freeze-thaw cycles as possible
- Does not contain insoluble material and is not colored or cloudy.
- Does not contain DNA. Always DNAse treat the RNA.
What elutant/suspension buffer should my sample be in?
HTSF prefers samples to be suspended in molecular biology grade water. Samples may also be suspended in different types of buffer, including 10mM Tris or Qiagen EB. DO NOT suspend samples in buffers containing EDTA.
Timeline
How long will it take to get my results?
Samples are processed on a first come first serve basis. Processing depends on the length of the queue. If you are submitting RNA or DNA, you should expect results in approximately 4 to 6 weeks, but we cannot guarantee this timeline. When submitting libraries or pools, your data will be delivered between 3-5 weeks, but we cannot guarantee this timeline.
If you have a deadline for a paper, a grant, or a presentation, please contact the project coordination team prior to submission. We will work with you to meet your desired deadline.
QAQC
What instruments are used to perform the quality check (QAQC)?
The HTSF uses a variety of QAQC instruments such as the TapeStation, LabChip, and Bioanalyzer to check RNA and DNA quality and the Qubit to assess RNA and DNA concentration.
How do I interpret my QAQC results?
Please visit the QAQC page tab for information on QAQC interpretation and common QAQC issues. The HTSF will contact you if your material fails QAQC and will provide information on how to move forward. The QAQC results can be found under “Attachments” on TracSeq.
Library Preparation
Does HTSF perform library preparation?
While we will accept user-made libraries, we can also construct libraries for a fee. Please note that we cannot guarantee successful sequencing of study-made libraries, and this is especially true for novel protocols. If you intend to perform a novel protocol, please contact Dr. Piotr Mieczkowski at piotr_mieczkowski@med.unc.edu. It is best to attach the protocol to your submission on TracSeq. If your protocol is custom, please select ‘Custom’ as your library preparation method in step 2 of the submission process. An empty cell will be provided for you to provide a brief description of your library preparation method.
What library preparation method should I use for my samples?
Your selected library preparation method will depend on your material type and project goals. Please contact the project coordination team to schedule a meeting to determine the best way to proceed with your samples. Additionally, a rough estimate of library type, sequencing depth, and sequencing methodology can be found on the Services page.
What are barcodes?
Unique “barcode” sequences are added to each DNA fragment during library preparation so that reads can be identified and sorted before data analysis. The choice of Illumina adapters depends on your library prep kit and application. Please reach out to the HTSF if you need help determining the best format.
How do I submit my barcodes/indexes correctly?
Submit indexes in the sample manifest as i7-i5/Index 1-Index 2 (ie. CCGCGGTT-AGCGCTAG). For i7 indexes, submit the barcode from the “sample sheet entry” column from the kit. For the i5, provide the forward strand workflow indexes.
Please contact htsf@med.unc.edu if you have any questions.
Sequencing
Which sequencing platform should I use?
The platform depends on the number of reads you require. The following chart will help you to determine which platform will best meet your data needs.
If you are unsure on how to proceed, please contact the project coordination team to schedule a consult to further discuss your sequencing plan.
What is the difference between the standard Novaseq and the NovaSeq XP?
The standard NovaSeq flowcell is designed to run a single pool across multiple lanes. The XP kit allows for running of multiple pools on a single flow cell. Below you will find detailed information regarding sequencing in standard vs. XP mode:
- Illumina NovaSeq Standard Loading Sequencing Platform Comparison Table
- Illumina NovaSeq XP Loading Sequencing Platform Comparison Table
What are the differences in data output between the HiSeq system and the NovaSeq?
As of Fall 2022, the HiSeq 2500/HiSeq 4000 sequencers are no longer in use. When changing from the HiSeq to the NovaSeq, HTSF found systematic bias; however, this switch did not broadly affect experimental results when evaluating mRNA-seq data. The magnitude of instrument bias is negligible relative to observed biological variation.
On the HTSF Landing page, What’s New section, there is a detailed review of the same libraries run on both platforms – PowerPoint HiSeq vs NovaSeq comparison.
I have custom sequencing primers which are required for my samples. How do I submit my primers?
During the submission process, you will have the option to select a custom sequencing primer. You will need to provide the primer name and sequence and bring custom primers with you at the time of drop-off. Information about submission requirements for custom sequencing primers can be accessed in Submitting Sequencing Custom Primers to HTSF.
If you require Old Nextera sequencing primer, please select this option during the submission process. The HTSF will supply the Old Nextera primer.
What is Single-end vs. paired-end reading?
During single-end sequencing, the library is only read in one direction. During paired-end sequencing, the library is read in both directions and provides twice as much sequencing data. Paired-end sequencing also detects insertions, deletions, or inversions.
What is sequencing coverage and sequencing depth?
Sequencing Coverage: The depth of coverage is a measure of the number of times that a specific genomic site is sequenced during a sequencing run. The higher the number of times that a base is sequenced, the better the quality of the data.
Sequencing Depth: We aim for a minimum of 30 million reads per sample for RNA sequencing. For more detailed RNA-seq analyses, such as allele-specific expression or expression of low-abundant transcripts, we recommend a greater read count.
What is cycle number (read length)?
Cycle number refers to the number of base pairs sequenced from an insert.
Why is PhiX added?
The HTSF adds the standard 1% PhiX that is required on all Illumina runs. If you suspect the complexity of the samples is low, additional PhiX may be required for the run to complete. See the Notes on PhiX Addition for Low Complexity Samples.
Consider your barcode selections prior to making your libraries if you are using dual barcodes. Do not select the same i7 barcodes for all libraries to be pooled and only vary the i5. It is best to have i7 vary so the sequencing run will be successful.
What happens to my samples after sequencing is complete?
The HTSF will store submitted samples for 1 year. After 1 year, the HTSF will contact you to determine if you want your material to be returned or discarded. Please contact the HTSF at any time for samples to be returned. The project coordination team is happy to arrange this for you.
Bioinformatics
Where can I find my data as an UNC member?
For UNC studies, a data folder will be established on the ITS Research Computing server. If you do not have a folder, the HTSF can request one at the time of setting up your account at the HTSF. The HTSF will put the data in the HTSF subfolder for your lab. You will receive an email when we place data in the folder at to completion of sequencing. If you can ot find the email or feel you data should have been delivered already, use the MY LANES feature on tracseq.
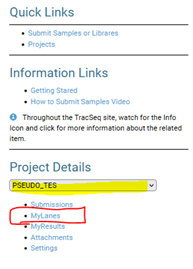
- Open Tracseq and on the right side you will find the Quick links
- Select your account and hit MYLANES below
- When MY LANES opens a new window, it will list all flowcells run for your account
- Use the name of your pool and the date to select the flowcell of interest (note the FC are listed from oldest to newest. Use the search filters)
- Hit the DETAILS button at the right
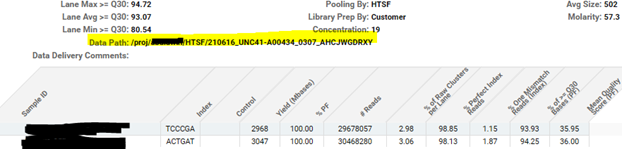
- This takes you to a flowcell summary with run set up, run metrics, demux data amounts for each library in your pool, data delivery folder info, etc.
- Data delivery folder information is located just above the pool break out for metrics. I will list the folder and the run name (highlighted below)
How do I access my data as an external account?
You will receive an email with a link to access your data. The data MUST be downloaded to your organization’s personal server within 5 days. After 5 days, the data is deleted. It can be reloaded for another 5 days at an additional charge. Please note that some sequencing platforms yield terabytes (Tb) of data and it can take some time for the download to be completed.
Can I get help analyzing my data?
Please reach out to our sister core, the Bioinformatics and Analytics Research Collaborative (BARC). For more information on BARC, please contact Dr. Hemant Kelkar at hkelkar@unc.edu.
Grants, Papers and Presentations
How do I confirm processing details for my paper?
Please contact the HTSF project coordination team @ htsf@med.unc.edu with the account name and batch number. We will confirm the work completed on your samples and will provide processing details.
How do I acknowledge the HTSF on my paper?
“We gratefully acknowledge the technical support from the UNC High Throughput Sequencing Facility. This facility is supported by the University Cancer Research Fund, Comprehensive Cancer Center Core Support grant (P30-CA016086) and the UNC Center for Mental Health and Susceptibility grant (P30-ES010126).”
What grant writing resources are available?