Examples
Examples of LiGaMD simulations
Trypsin-Benzamidine Binding:
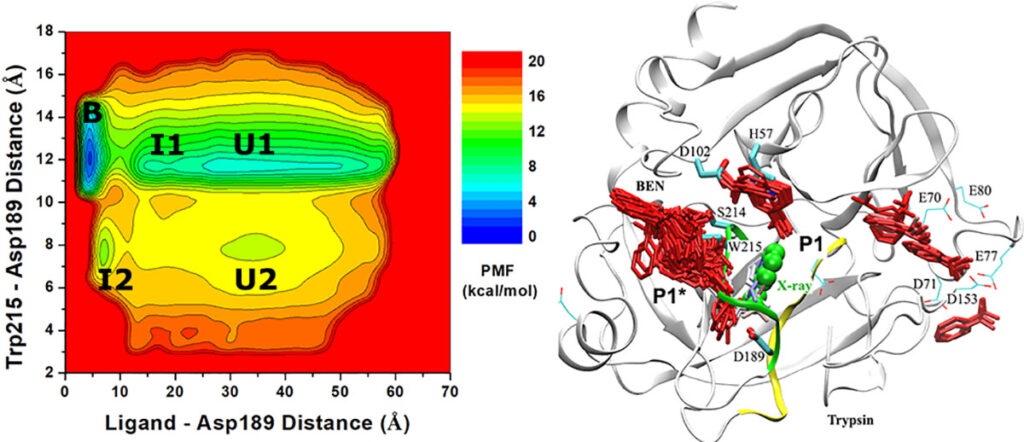
All-atom ligand Gaussian accelerated molecular dynamics (LiGaMD) simulations captured repeptitive binding of the benzamidine (BEN) ligand to the trypsin model protein, which enabled us to characterize the ligand binding free energy profiles, pathways and kinetic rate constants.
Simulation Movie: Trypsin-benzamidine Sim2
Ligand Gaussian accelerated Molecular Dynamcis (LiGaMD) simulation of repetitive dissociation and binding of a ligand molecule benzamidine to target protein trypsin.
Simulation Movie: Trypsin-benzamidine Sim5
Ligand Gaussian accelerated Molecular Dynamcis (LiGaMD) simulation of repetitive dissociation and binding of a ligand molecule benzamidine to target protein trypsin.
Reference: Miao, Y.; A. Bhattarai; and J. Wang, Ligand Gaussian accelerated molecular dynamics (LiGaMD): Characterization of ligand binding thermodynamics and kinetics. Journal of Chemical Theory and Computation, 16(9): 5526-5547.
Examples of GaMD simulations
Protein folding:

Folding of chignolin was captured in GaMD simulations: the unfolded (“U”), intermediate (“I”) and folded (“F”) states (blue) aligned to the PDB native structure (red). Residues including Tyr2, Asp3, Pro4, Glu5, Thr6, Thr8 and Trp9 are represented by sticks. Notably, Trp9 and Pro4 form hydrophobic interactions in the intermediate state.
Simulation Movie: Chignolin Folding
Reference: Miao, Y.; Feher, V. A.; McCammon, J. A., Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J Chemical Theory and Computation, 11(8): 3584-3595.
Protein-ligand Binding:

A pathway of benzene binding to the T4-lysozyme was observed during the GaMD simulation. The unbound (“U”), intermediate (“I”) and bound (“B”) poses of the protein-ligand complex (blue) with the protein C-terminal domain (residues 80-160) aligned to the PDB native structure (red). The protein and benzene are represented by ribbons and spheres, respectively, and they are colored by blue for the simulation structure while red for the PDB native structure, except that in the binding pathway the simulated benzene is represented by lines and colored by simulation time in a BWR color scale. Residues with heavy atoms found within 3 Å of benzene are represented by sticks.
Simulation Movie: T4-lysozyme ligand binding
GaMD simulation of ligand binding to T4 lysozyme
Reference: Miao, Y.; Feher, V. A.; McCammon, J. A., Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J Chemical Theory and Computation, 11(8): 3584-3595.
Graded Activation and Free Energy Landscapes of a Muscarinic G-Protein-Coupled Receptor:
G-protein–coupled receptors (GPCRs) represent primary targets of about one-third of currently marketed drugs. The structure, dynamics, and function of GPCRs result from complex free energy landscapes. In this work, we have applied Gaussian accelerated molecular dynamics (GaMD) to study the ligand-dependent behavior of the M2 muscarinic GPCR. Extensive GaMD simulations have revealed distinct structural flexibility and free energy profiles that depict graded activation of the M2 receptor. We have captured both dissociation and binding of an orthosteric ligand in a single all-atom GPCR simulation. GaMD is well poised to study large biomolecules and ligand recognition for drug discovery.
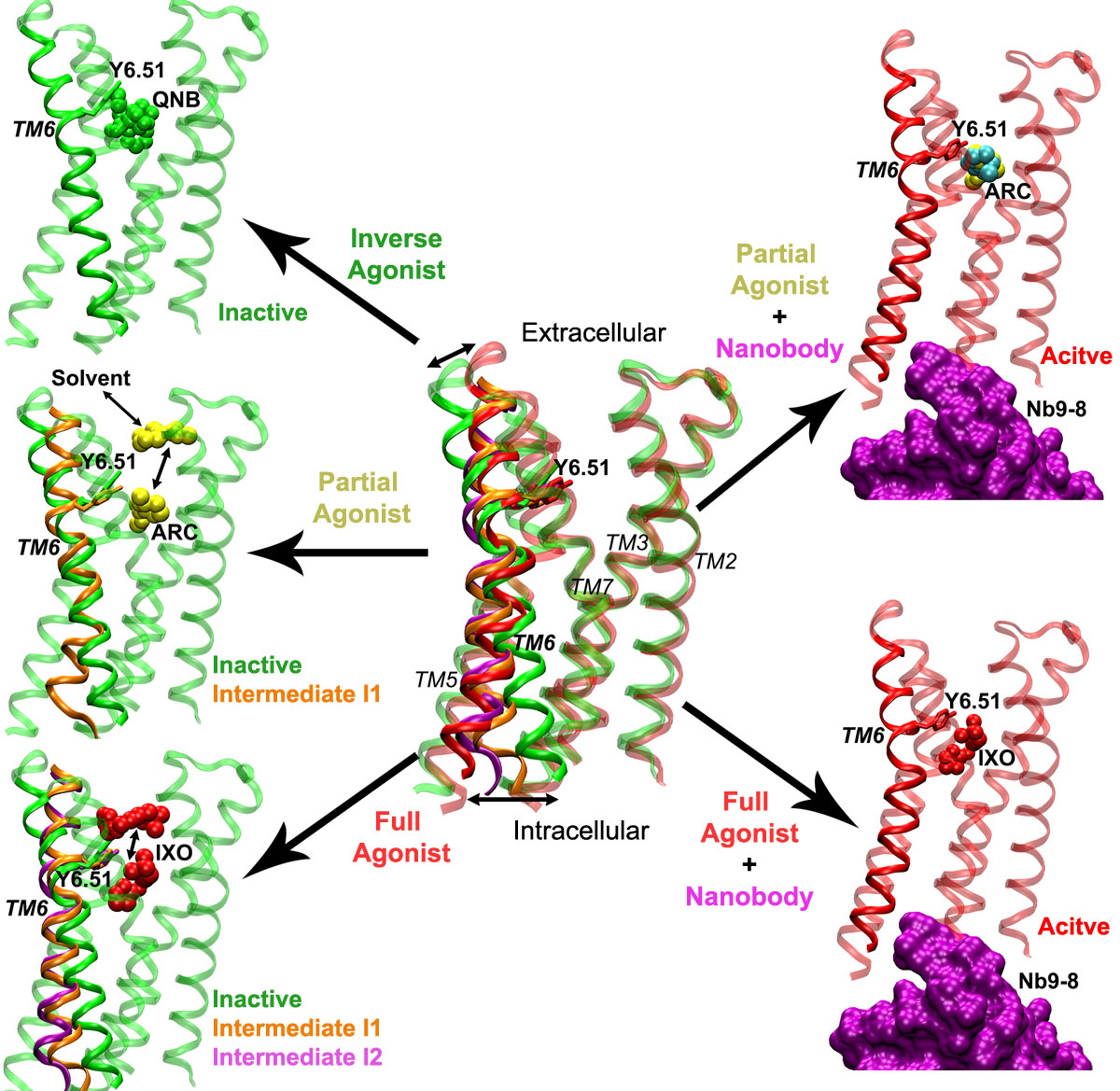
The M2 receptor (ribbons) samples a large conformational space with significant structural rearrangements, especially for the TM6 helix. Binding of the inverse agonist QNB (green spheres) confines the receptor in the inactive state. Without the G protein or mimetic nanobody, the partial agonist ARC (yellow spheres) biases the M2 receptor to visit an intermediate state “I1” (orange ribbons). ARC is able to dissociate completely to the bulk solvent via the extracellular vestibule and rebinds to the receptor repeatedly during a 2030 ns GaMD simulation. In comparison, the full agonist IXO (red spheres) biases the receptor further, sampling both intermediate “I1” (orange ribbons) and “I2” (purple ribbons). IXO escapes out of the orthosteric pocket and visits the extracellular vestibule in one of the GaMD simulations. By adding the G protein mimetic nanobody (purple surface), the M2 receptor is stabilized in the fully active state (red ribbons) as bound by IXO or ARC, although ARC adopts two alternative conformations in the orthosteric pocket, ARC-P1 (yellow spheres) and ARC-P1′ (cyan spheres).

Pathways of partial agonist dissociation and binding observed in GaMD simulation. (A) Timecourse of the ARC?Asp1033.32 distance during 2030 ns simulation. Four dissociation and three binding events are labeled. (B-H) Schematic representations of the ligand pathways during (B) “D1”, (C) “B1”, (D) “B2”, (E) “B3”, (F) “D2”, (G) “D3” and (H) “D4”. The receptor is represented by blue ribbons and the ligand by sticks colored by the position along the membrane normal. (I) Ten lowest energy structural clusters of ARC that are labeled and colored in a GWR scale according to the PMF values.
Simulation Movie: M2-ARC dissociation and binding
The dissociation and binding of partial agonist ARC observed in GaMD simulation of the M2-ARC complex system. Not only does ARC escape out of the receptor orthosteric pocket, but it also dissociates completely and rebinds multiple times to the receptor during the 2030 ns GaMD simulation.
Reference: Y. Miao and J. A. McCammon (2016), Graded activation and free energy landscapes of a muscarinic G protein-coupled receptor. Proc Natl Acad Sci U S A, 113 (43): 12162–12167.