Theory
Download a PDF copy of GaMD theory.
1. Gaussian accelerated molecular dynamics (GaMD)
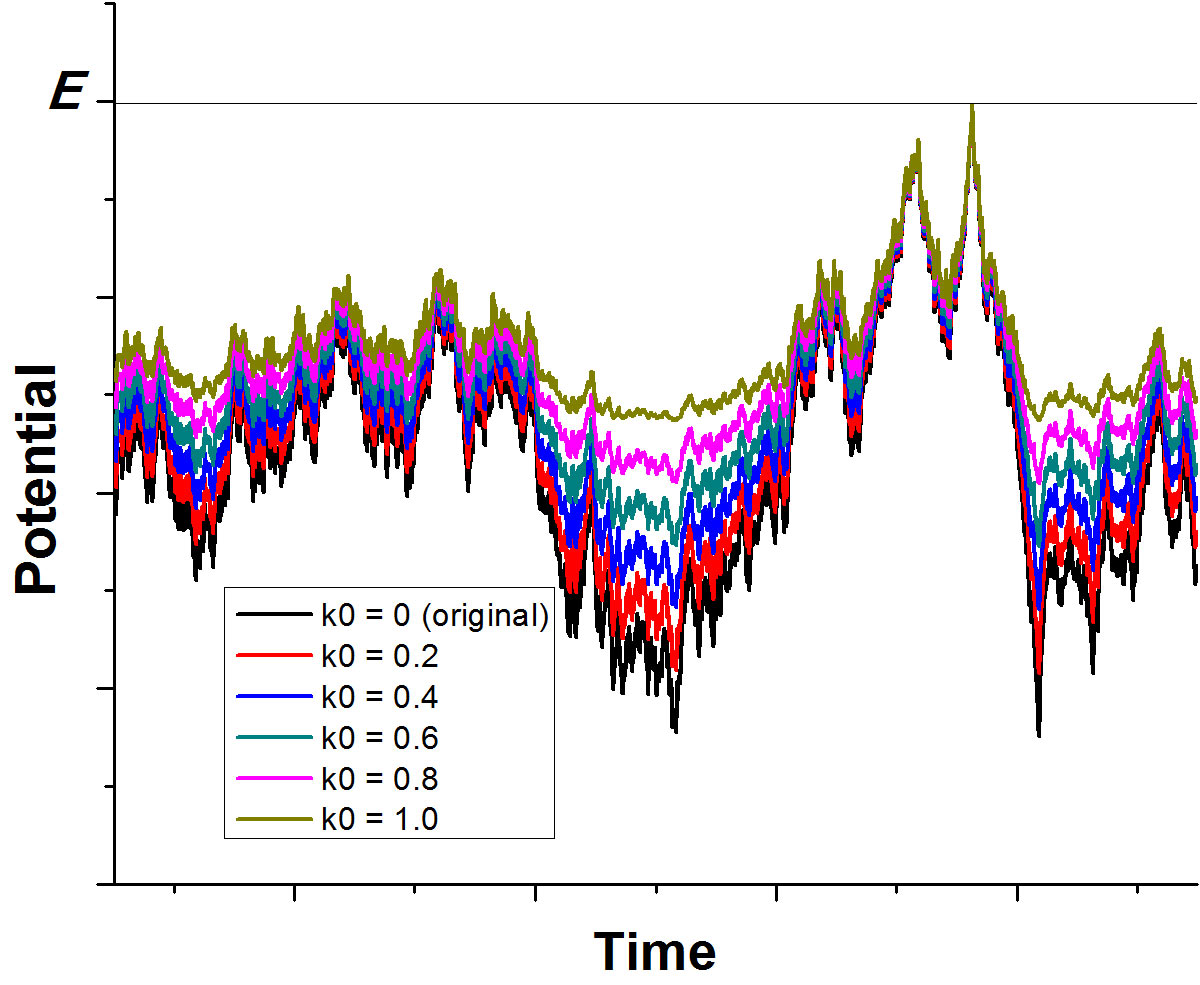
Gaussian Accelerated Molecular Dynamics (GaMD) has been developed for simultaneous unconstrained enhanced sampling and free energy calculation of large biomolecules. By constructing a boost potential that follows Gaussian distribution, accurate reweighting of the GaMD simulations is achieved using cumulant expansion to the second order. GaMD has been demonstrated on three biomolecular model systems: alanine dipeptide, chignolin folding and ligand binding to the T4-lysozyme. Without the need to set predefined reaction coordinates, GaMD enables unconstrained enhanced sampling of these biomolecules. Furthermore, the free energy profiles obtained from reweighting of the GaMD simulations allow us to identify distinct low energy states of the biomolecules and characterize the protein folding and ligand binding pathways quantitatively.
Reference
Miao, Y.*, Feher V. and McCammon JA (2015) Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J Chemical Theory and Computation, 11(8): 3584-3595.
2. Ligand Gaussian accelerated molecular dynamics (LiGaMD)
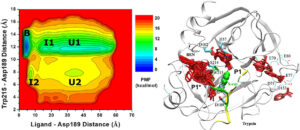
Based on GaMD, a new algorithm called Ligand GaMD or “LiGaMD” has been developed to simulate ligand binding and unbinding. It works by selectively boosting the ligand non-bonded interaction potential energy. Another boost potential could be applied to the remaining potential energy of the entire system in a dual-boost algorithm (LiGaMD_Dual) to facilitate ligand binding. LiGaMD has been demonstrated on host-guest and protein-ligand binding model systems. Repetitive guest binding and unbinding in the β-cyclodextrin host were observed in hundreds-of-nanosecond LiGaMD simulations. The calculated binding free energies of guest molecules with sufficient sampling agreed excellently with experimental data (< 1.0 kcal/mol error). In comparison with previous microsecond-timescale conventional molecular dynamics simulations, accelerations of ligand kinetic rate constants in LiGaMD simulations were properly estimated using Kramers’ rate theory. Furthermore, LiGaMD allowed us to capture repetitive dissociation and binding of the benzamidine inhibitor in trypsin within 1 μs simulations. The calculated ligand binding free energy and kinetic rate constants compared well with the experimental data. Therefore, LiGaMD provides a promising approach for characterizing ligand binding thermodynamics and kinetics simultaneously.
Reference
Miao, Y.; A. Bhattarai; and J. Wang, (2020) Ligand Gaussian accelerated molecular dynamics (LiGaMD): Characterization of ligand binding thermodynamics and kinetics. Journal of Chemical Theory and Computation, 16(9): 5526-5547.
3. Peptide Gaussian accelerated molecular dynamics (Pep-GaMD)
Peptides mediate up to 40% of known protein-protein interactions in higher eukaryotes and play an important role in cellular signaling. However, it is challenging to simulate both binding and unbinding of peptides and calculate peptide binding free energies through conventional molecular dynamics, due to long biological timescales and extremely high flexibility of the peptides. Based on the GaMD enhanced sampling technique, we have developed a new computational method “Pep-GaMD”, which selectively boosts essential potential energy of the peptide in order to effectively model its high flexibility. In addition, another boost potential is applied to the remaining potential energy of the entire system in a dual-boost algorithm. Pep-GaMD has been demonstrated on binding of three model peptides to the SH3 domains. Independent 1 μs dual-boost Pep-GaMD simulations have captured repetitive peptide dissociation and binding events, which enable us to calculate peptide binding thermodynamics and kinetics. The calculated binding free energies and kinetic rate constants agreed very well with available experimental data. Furthermore, the all-atom Pep-GaMD simulations have provided important insights into the mechanism of peptide binding to proteins that involves long-range electrostatic interactions and mainly conformational selection. In summary, Pep-GaMD provides a highly efficient, easy-to-use approach for unconstrained enhanced sampling and calculations of peptide binding free energies and kinetics.
Reference
Wang, J. and Miao, Y*, (2020) Peptide Gaussian accelerated molecular dynamics (Pep-GaMD): Enhanced sampling and free energy and kinetics calculations of peptide binding. Journal of Chemical Physics, 153, 154109.
4. Protein-protein interaction-Gaussian accelerated molecular dynamics (PPI-GaMD)
Protein-protein interactions (PPIs) play key roles in many fundamental biological processes such as cellular signaling and immune responses. However, it has proven challenging to simulate repetitive protein association and dissociation in order to calculate binding free energies and kinetics of PPIs, due to long biological timescales and complex protein dynamics. To address this challenge, we have developed a new computational approach to all-atom simulations of PPIs based on GaMD. The method, termed “PPI-GaMD”, selectively boosts interaction potential energy between protein partners to facilitate their slow dissociation. Meanwhile, another boost potential is applied to the remaining potential energy of the entire system to effectively model the protein’s flexibility and rebinding. PPI-GaMD has been demonstrated on a model system of the ribonuclease barnase interactions with its inhibitor barstar. Six independent 2 μs PPI-GaMD simulations have captured repetitive barstar dissociation and rebinding events, which enable calculations of the protein binding thermodynamics and kinetics simultaneously. The calculated binding free energies and kinetic rate constants agree well with the experimental data. Furthermore, PPI-GaMD simulations have provided mechanistic insights into barstar binding to barnase, which involve long-range electrostatic interactions and multiple binding pathways, being consistent with previous experimental and computational findings of this model system. In summary, PPI-GaMD provides a highly efficient and easy-to-use approach for binding free energy and kinetics calculations of PPIs.
Reference
Wang, J. and Miao, Y*, (2022) Protein-protein interaction-Gaussian accelerated molecular dynamics (PPI-GaMD): Characterization of protein binding thermodynamics and kinetics. Journal of Chemical Theory and Computation, 18(3):1275-1285.