
3 weeks ago

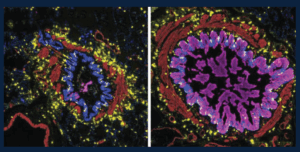
Many of the small airways in severe asthma and from people who die from asthma contain mucus that blocks the airways and prevents air from flowing normally when these patients breathe. This figure shows the variety of patterns in mucus in the small airways: those that are blocked with lots of mucus in the epithelial cells (magenta, right) and those that are not (left). The immune cells in the lungs, including a special type of allergy-associated cell (mast cells, yellow), are distributed equally between the mucus-containing airways and those airways without mucus. These studies help us begin to understand why certain airways become filled with mucus, which can help us to find treatments targeting mucus in asthma.
Credit: Schworer SA*, Murano H*, Dang H, Markovetz MR, Saito M, Kato T, Asakura T, Chen G, Gilmore RC, Morton LC, van Heusden C, Chua M, Strickler E, Wisniewski ZY, Crisp G, Mitchell E, Doherty KA, Mastan S, Trejo Bittar HE, Cody BA, Trudeau JB, De la Cruz G, Ralph LM, Askin FB, Panettieri RA Jr, Koziol-White CJ, Byrd KM, Livraghi-Butrico A, O’Neal WK, Randell SH, Wenzel SE#, Okuda K#, Boucher RC Jr#. Airway epithelial heterogeneity and mucus plugging in asthmatic bronchioles. Am J Respir Crit Care Med. 2026 Feb;212(2):209-226. doi: 10.1164/rccm.202409-1849OC. PMID: 40986379. PMCID: PMC12668797. *Co-First Authors. #Co-Senior Authors.
The image was provided by Stephen Schworer, MD, PhD, and was featured on the cover of the February 2026 issue of American Journal of Respiratory and Critical Care Medicine.
3 weeks ago

4 weeks ago

1 month ago

2 months ago

2 months ago

3 months ago